Ian Michelow, et al. : Griffithsin vs Ebola Virus -- Articles
& patents
**
[rexresearch.com](../index.htm)**
---
**Ian Michelow*, et al.*
Griffithsin vs Ebola Virus**
---
**---
**[Case Adams : Red
Algae Extract Fights Ebola ... and HIV, SARS and HCV](#redalgae)****[High-Dose Mannose-Binding
Lectin Therapy for Ebola Virus Infection](#J_Infect_Dis)****[Isolation and characterization
of griffithsin](#isolation)****[US2010331240 : METHODS FOR
PREVENTION AND TREATMENT OF INFECTIONS WITH
SUPRAPHYSIOLOGICAL DOSES OF MANNAN-BINDING LECTIN](#US2010331240_)****[US8394764 : GRIFFITHSIN,
GLYCOSYLATION-RESISTANT GRIFFITHSIN, AND RELATED
CONJUGATES,](#US8394764)****[US8088729 : Anti-viral
griffithsin compounds, compositions, and methods of use](#US8088729)****[Scytonema varium red algae /
Scytovirin Patents](#Scytovirin)**[**Nostoc ellipsosporum /
Cyanovirin-N Patents**](#Cyanovirin-N)**
**---
[**http://www.greenmedinfo.com/blog/red-algae-extract-fights-ebola-and-hiv-sars-and-hcv**](http://www.greenmedinfo.com/blog/red-algae-extract-fights-ebola-and-hiv-sars-and-hcv)
**Red Algae Extract Fights Ebola
... and HIV, SARS and HCV**
**by** **Case Adams**** Ebola Antibodies
The research found that nearly half of those who were asymptomatic
and seemingly immune developed antibodies (IgM and IgG) to the
Ebola virus.
This means these individuals certainly were intimately exposed to
the virus, but simply naturally developed the immunity tools -
including those discussed below - that prevented the infection
from replicating out of control.
Furthermore, the asymptomatic group exhibited greater
anti-inflammatory responses in general. They were found to have
higher levels of circulating cytokines and chemokines a which
speed up the body's natural ability to break down the viral cells
and stop their activity within the body.
They concluded: "Asymptomatic individuals had a strong
inflammatory response by high circulating concentrations of
cytokines and chemokines."
Mannose-Binding Lectins Attack Ebola Virus
The particular mechanism with which the body naturally breaks down
and prevents infection from lethal infections including Ebola,
HIV, HCV and SARS has gradually emerged.
The mechanism is called mannose-binding lectins. Mannose-binding
lectins are apparently produced in the human body via a DNA
sequence, called the MBL2.
When this part of our genes is in order, the body will produce and
release these mannose-binding lectins into the bloodstream.
Mannose-binding lectins will then recognize and glom onto certain
carbohydrate molecules that cover and make up various
microorganisms.
These include fungi, bacteria and even parasites, which utilize
glycoprotein shells to protect themselves. But they also include
viruses. Once the lectins attach to these shells, they will break
apart the surface of the microbe and basically break them down,
allowing the body's other immune cells to kill off the microbe and
prevent it from replicating.
In fact, a healthy body that produces good levels of these
mannose-binding lectins will be able to easily fight off colds and
flus, as well as other microbial infections. Several animal
studies have shown mannose-binding lectins heartily beat down
coronaviruses and infectious bronchitis.
Research over the past five years has found that low levels of
mannose-binding lectins increases the risk of respiratory
infections, including syncytial virus infections, pneumonia and
others.
For example, in a study of 121 children, RSV-infections were
associated with low levels of mannose-binding lectins. Nearly 70
percent of RSV-infected children had low levels of mannose-binding
lectins. But other infections a especially those related to
bacterial infections a are not necessarily connected with
mannose-binding lectin levels.
When it comes to virulent infections such as Ebola, Hepatitis C
and HIV, however, these are different. These viruses come with
glycoprotein shells that protect the virus from being broken down.
Furthermore, the glycoprotein shell of the Ebola virus produces
glycoproteins that damage cells, allowing the virus to penetrate
and replicate within the cell.
Mannose-binding lectins actually break down this shell and the
glycoprotein matrix through a mechanism called the lectin pathway.
Humans that don't produce enough of these mannose-binding lectins
are not only more susceptible because they don't have enough
lectins, but they are typically also immunosuppressed with regard
to the rest of their immune system.
One of the reason some humans don't produce enough mannose-binding
lectins is because of a slight genetic mutation, where the MBL2
gene is switched off. The reason for this mutation/switch-off has
yet to be fully understood. (Guess - something to do with our
toxic environment and/or nutritional deficiency.)
Mannose-Binding Lectins From Red Algae
This brings us to the fun part. Yes, humans aren't the only
critters that produce mannose-binding lectins. Red algae also
produce these profusely, which allow the algae to protect
themselves from invasion by viruses.
The most promising form of mannose-binding lectins is a component
of the Scytonema varium red algae called Scytovirin. The protein
extract was isolated by researchers from the National Cancer
Institute at Frederick, Maryland in 2003. The protein contains 95
amino acids, and was found to bind to HIV-1 viral shells.
A similar antiviral protein was found in Nostoc ellipsosporum a
called Cyanovirin-N. Both of these antiviral proteins did similar
things a they broke down the glycoprotein shells of HIV and HCV.
Yet another anti-viral extract was found from the New Zealand red
alga species, Griffithsia sp. This protein is called Griffithsin,
abbreviated with GRFT.
Over the next few years, Griffithsin was tested against HIV-1 with
great success in laboratory studies, which included studies with
mice. The epidemic-potential virus SARS was also tested against
Griffithsin, also with great success.
Multiple studies illustrated these effects. Research from the
Center for Cancer Research in Frederick, Maryland found that
Griffithsin not only stopped HIV-1 virus replication, but stopped
cellular intrusion of the virus.
In 2010 Harvard researchers tested a recombinant version of
Griffithsin a called rhMBL a against Ebola. Once again, they found
the mannose-binding lectins were able to not only breakdown the
viral shells of the Ebola, but when given to mice infected with
Ebola, the mice became immune to the virus.
Yes, when the mice given the recombinant mannose-binding lectins
were rechallenged with the Ebola virus, they were found to be
immune to the Ebola virus.
Since that study other research has tested other animals with
Griffithsin, with similar results.
Recombinant Griffithsin Produced in Nicotiana Benthamiana Plants
As modern medical researchers continually strive for isolated and
synthesized versions of nature able to be patented, recombinant
versions of Griffithsin were eventually produced using Nicotiana
benthamiana plants (a relative of the tobacco plant). These plants
were genetically modified so they would produce the same
mannose-binding lectins.
This form of Griffithsin was tested on mice and guinea pigs
infected with HIV-1, with successful antiviral results.
This was also found when testing the recombinant Griffithsin on
Ebola-infected mice.
In all the studies, the Griffithsin was found to be safe and
tolerated.
As to whether red algae can be taken in natural form to increase
immunity, there is no doubt this is the case. Prior to this
antiviral research that has spiraled into biopharm research, red
algae had been shown to have antiviral and anticancer effects.
So the most logical answer is "yes" a certainly consuming red
algae in supplement form has been found to boost antiviral
immunity, and from the available research, blood levels of
mannose-binding lectins. This should in turn boost immunity and
create a natural method of preventing and even treating viral
infections such as Ebola, SARS, HIV and Hepatitis-C.
Of course, this strategy should be used with other natural
immunity-boosting strategies.
Other plants also produce these mannose-binding lectins, some of
which have been used in traditional medicines. A study from
Belgium's University of Leuven studied 33 different plant lectins,
and found 10 different mannose-binding lectins among the plants
that inhibited coronovirus, and intervened upon the replication
cycle of SARS-CoV.
Consult with your health professional if you are sick.
REFERENCES:
Baize S, Leroy EM, Georges-Courbot MC, Capron M, Lansoud-Soukate
J, DebrA(c) P, Fisher-Hoch SP, McCormick JB, Georges AJ. Defective
humoral responses and extensive intravascular apoptosis are
associated with fatal outcome in Ebola virus-infected patients.
Nat Med. 1999 Apr;5(4):423-6.
http://www.ncbi.nlm.nih.gov/pubmed/10202932
http://www.nature.com/nm/journal/v5/n4/full/nm0499\_423.html
Leroy EM, Baize S, Volchkov VE, Fisher-Hoch SP, Georges-Courbot
MC, Lansoud-Soukate J, Capron M, DebrA(c) P, McCormick JB, Georges
AJ. Human asymptomatic Ebola infection and strong inflammatory
response. Lancet. 2000 Jun 24;355(9222):2210-5.
http://www.ncbi.nlm.nih.gov/pubmed/10881895
http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(00)02405-3/fulltext
Albert RK, Connett J, Curtis JL, Martinez FJ, Han MK, Lazarus SC,
Woodruff PG. Mannose-binding lectin deficiency and acute
exacerbations of chronic obstructive pulmonary disease. Int J
Chron Obstruct Pulmon Dis. 2012;7:767-77. doi:
10.2147/COPD.S33714.
http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(00)02405-3/fulltext
http://www.dovepress.com/mannose-binding-lectin-deficiency-and-acute-exacerbations-of-chronic-o-peer-reviewed-article-COPD
Ribeiro LZ, Tripp RA, Rossi LM, Palma PV, Yokosawa J, Mantese OC,
Oliveira TF, Nepomuceno LL, QueirA3z DA. Serum mannose-binding
lectin levels are linked with respiratory syncytial virus (RSV)
disease. J Clin Immunol. 2008 Mar;28(2):166-73.
http://www.ncbi.nlm.nih.gov/pubmed/17952574
http://link.springer.com/article/10.1007%2Fs10875-007-9141-8
Barton C, Kouokam JC, Lasnik AB, Foreman O, Cambon A, Brock G,
Montefiori DC, Vojdani F, McCormick AA, O'Keefe BR, Palmer KE.
Activity of and effect of subcutaneous treatment with the
broad-spectrum antiviral lectin griffithsin in two laboratory
rodent models. Antimicrob Agents Chemother. 2014;58(1):120-7. doi:
10.1128/AAC.01407-13.
http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0064449
http://aac.asm.org/content/58/1/120.long
Takebe Y, Saucedo CJ, Lund G, Uenishi R, Hase S, Tsuchiura T,
Kneteman N, Ramessar K, Tyrrell DL, Shirakura M, Wakita T, McMahon
JB, O'Keefe BR. Antiviral lectins from red and blue-green algae
show potent in vitro and in vivo activity against hepatitis C
virus. PLoS One. 2013 May 21;8(5):e64449. doi:
10.1371/journal.pone.0064449.
http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0064449
http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0064449
Mori T, O'Keefe BR, Sowder RC 2nd, Bringans S, Gardella R, Berg S,
Cochran P, Turpin JA, Buckheit RW Jr, McMahon JB, Boyd MR.
Isolation and characterization of griffithsin, a novel
HIV-inactivating protein, from the red alga Griffithsia sp. J Biol
Chem. 2005 Mar 11;280(10):9345-53.
http://www.ncbi.nlm.nih.gov/pubmed/15613479
http://www.jbc.org/content/280/10/9345.long
Bokesch HR, O'Keefe BR, McKee TC, Pannell LK, Patterson GM,
Gardella RS, Sowder RC 2nd, Turpin J, Watson K, Buckheit RW Jr,
Boyd MR. A potent novel anti-HIV protein from the cultured
cyanobacterium Scytonema varium.
Biochemistry. 2003 Mar 11;42(9):2578-84.
http://www.ncbi.nlm.nih.gov/pubmed/12614152
http://pubs.acs.org/doi/abs/10.1021/bi0205698
Michelow IC, Lear C, Scully C, Prugar LI, Longley CB, Yantosca LM,
Ji X, Karpel M, Brudner M, Takahashi K, Spear GT, Ezekowitz RA,
Schmidt EV, Olinger GG.
High-dose mannose-binding lectin therapy for Ebola virus
infection.
J Infect Dis. 2011 Jan 15;203(2):175-9. doi:
10.1093/infdis/jiq025.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3071052/
http://jid.oxfordjournals.org/content/203/2/175.long
Vorup-Jensen T, SA,rensen ES, Jensen UB, Schwaeble W, Kawasaki T,
Ma Y, Uemura K, Wakamiya N, Suzuki Y, Jensen TG, Takahashi K,
Ezekowitz RA, Thiel S, Jensenius JC.
Recombinant expression of human mannan-binding lectin.
Int Immunopharmacol. 2001 Apr;1(4):677-87.
http://www.ncbi.nlm.nih.gov/pubmed/11357880
http://www.sciencedirect.com/science/article/pii/S1567576900000527
Singh RS, Thakur SR, Bansal P.
Algal lectins as promising biomolecules for biomedical research.
Crit Rev Microbiol. 2013 Jul 16.
http://www.ncbi.nlm.nih.gov/pubmed/23855360
http://informahealthcare.com/doi/abs/10.3109/1040841X.2013.798780
Keyaerts E, Vijgen L, Pannecouque C, Van Damme E, Peumans W,
Egberink H, Balzarini J, Van Ranst M.
Plant lectins are potent inhibitors of coronaviruses by
interfering with two targets in the viral replication cycle.
Antiviral Res. 2007 Sep;75(3):179-87.
http://www.ncbi.nlm.nih.gov/pubmed/17428553
http://www.sciencedirect.com/science/article/pii/S0166354207002380
While researchers scramble to develop a vaccine or monoclonal
antibody against the Ebola virus a and continue to develop chemo
treatments to stem HIV and Hepatitis-C while fearing SARS a nature
has already provided a natural treatment.
Research has shown that a healthy strong immune system can allow a
person to not only avoid contracting the disease a but become
resistant to it as well.
For those of us who need help or extract assurance, red algae
proves to provide a key antiviral.
Hunting Natural Immunity For Ebola
After the two 1996 Ebola outbreaks in Gabon Africa, medical
scientists determined that about Ebola causes death among about 70
percent of those who contracted the virus.
This question led researchers from Gabon's Franceville
International Center of Medical Research to investigate. The
questions ensued: Why don't the other 30 percent die? How do 30
percent of those infected recover?
Furthermore, medical researchers found many instances where there
were close contacts of those who became infected who never were
infected at all. Even though they were in contact with the
infected patient while the patient was symptomatic.
Note: An infected patient with Ebola must be symptomatic in order
to be contagious a with fever and other flu-like symptoms. A
person must also have direct mucosal or blood contact in order to
become infected with the virus. This means a transfer of saliva,
urine, semen or blood from one person to another.
Thus, when the researchers investigated "close contact"
individuals, they focused upon those who had this sort of
exposure.
**---
**Griffithsia species**
  
---
**J Infect Dis. 2011 Jan 15;203(2):175-9. doi:
10.1093/infdis/jiq025.** **** ****
**High-Dose Mannose-Binding Lectin
Therapy for Ebola Virus Infection**
Ian C. Michelow,1 Calli Lear,2 Corinne Scully,2 Laura I.
Prugar,2 Clifford B. Longley,3 L. Michael Yantosca,1 Xin Ji,4
Marshall Karpel,1 Matthew Brudner,1 Kazue Takahashi,1 Gregory T.
Spear,4 R. Alan B. Ezekowitz,1 Emmett V. Schmidt,corresponding
author5 and Gene G. Olinger2
**Abstract**
Mannose-binding lectin (MBL) targets diverse microorganisms for
phagocytosis and complement-mediated lysis by binding specific
surface glycans. Although recombinant human MBL (rhMBL) trials
have focused on reconstitution therapy, safety studies have
identified no barriers to its use at higher levels. Ebola
viruses cause fatal hemorrhagic fevers for which no treatment
exists and that are feared as potential biothreat agents. We
found that mice whose rhMBL serum concentrations were increased
=7-fold above average human levels survived otherwise fatal
Ebola virus infections and became immune to virus rechallenge.
Because Ebola glycoproteins potentially model other glycosylated
viruses, rhMBL may offer a novel broad-spectrum antiviral
approach.
Circulating mannose-binding lectin (MBL) is a first-line host
defense against a wide range of viral and other pathogens. MBL
is a C-type lectin that recognizes hexose sugars including
mannose, glucose, fucose, and N-acetylglucosamine on the surface
of many pathogens. It does not recognize the terminal
carbohydrates galactose and sialic acid on normal host cells.
Therefore, MBL preferentially recognizes glycosylated viruses
including influenza virus, human immunodeficiency virus, severe
acute respiratory syndrome coronovirus (SARS-CoV), Ebola virus,
and Marburg virus. It also recognizes many glycosylated
gram-positive and gram-negative bacteria [1, 2]. As a result of
common genetic variants, MBL serum levels in humans range from 0
to 10,000 ng/mL. Thirty percent of the human population has
levels <500 ng/mL, which are associated with increased
susceptibility to infections in children and immunocompromised
individuals [3].
We previously reported preclinical studies that addressed the
potential utility of recombinant human MBL (rhMBL)
reconstitution therapy. MBL-knockout mice are highly susceptible
to several bacteria including Staphylococcus aureus [1]. RhMBL
improved survival in MBL-null mice to approximate survival among
infected wild-type mice at doses that reconstituted the
complement-activating capacity of MBL-knockout serum to a level
comparable to that of wild-type mouse serum [1]. Doses of
plasma-derived MBL and rhMBL designed to increase MBL
concentrations to physiologic levels (>1000 ng/mL) in
MBL-deficient humans were safe in early trials and did not
elicit antibodies [3a5]. In contrast, although MBL replacement
therapy enhanced opsonophagocytic potential, higher levels of
plasma-derived MBL were needed to achieve MBL-mediated
complement activation comparable to healthy controls [6],
suggesting that above-replacement dosing will need attention.
Ebola and Marburg viruses of the filovirus family are among the
most virulent causes of the human viral hemorrhagic fevers and
cause devastating epidemics of fulminant and rapidly fatal
disease. They constitute important biological threat agents
because of their high mortality rates, capacity for large-scale
dissemination, and potential for causing social disruption.
Currently, there are no US Food and Drug Administrationaapproved
therapeutic agents available to prevent or treat these lethal
viral infections. Filovirus surface glycoproteins (GPs) are
heavily glycosylated and contain high-mannose. As a result, MBL
binds to Ebola and Marburg viruses and mediates
complement-dependent virus neutralization [2]. Importantly,
their surface glycoprotein structures are characteristic of a
broad group of viruses in which N-linked glycosylation
contributes to viral virulence [7]. Reasoning that MBL treatment
is likely to be safe at supraphysiological levels, we evaluated
an in vivo Ebola virus model to explore the possibility of using
MBL as an immunotherapeutic agent. Our results showed that
supraphysiological doses of MBL rescued ~40% of mice from lethal
challenges when administered prea or postaEbola virus exposure.
This novel paradigm suggests that high-dose MBL should be
evaluated more broadly as an immunotherapeutic agent for a wide
spectrum of glycosylated pathogens.
**MATERIALS AND METHODS**
**Production and pharmacokinetics of rhMBL**
Commercial-grade rhMBL was provided by Enzon Pharmaceuticals
[8]. Human MBL concentrations and complement cleavage activity
were measured as described elsewhere [9]. Pharmacokinetics of
rhMBL concentrationatime data were evaluated using
noncompartmental modeling with WinNonlin Professional Edition
(version 5.2; Pharsight). The area under the curve from zero to
infinity (AUC0a8) values were calculated using the linear
trapezoidal method.
**Murine Ebola model**
We used a validated lethal Ebola Zaire mouse model developed at
the US Army Medical Research Institute of Infectious Diseases
(USAMRIID) [10], with a double plaque-purified, mouse-adapted,
Ebola isolate, EZ'76 Mp3 Vp2 Mp9 GH. The virus was inoculated
intraperitoneally (i.p.) at 100 pfu (3000 A LD50) producing
uniformly lethal disease in C57B6 mice using biosafety level-4
facilities. Research was conducted in compliance with the Animal
Welfare Act and federal regulations in a fully accredited
facility. To assess the effect of rhMBL on virus lethality, we
treated Ebola virusainfected C57B6 mice i.p. with either 4.3
mg/kg or 20 mg/kg of rhMBL twice daily ~12 hours apart for 10
days. On the day of virus exposure, mice were treated and
exposed to 100 pfu of mouse-adapted Ebola Zaire either 12 hours
before or 1 hour after the first dose of rhMBL as indicated in
Figure 1.
**Figure 1.****Survival and laboratory indices of filovirus-infected
mice treated with recombinant human mannose-binding lectin
(rhMBL). (A) Mouse survival when treated with rhMBL before
Ebola virus inoculation. Sham-treated wild-type mice were
compared with wild-type ...**
Mice were assessed daily for changes in physical appearance and
weight. Viremia was assessed by reverse transcription-polymerase
chain reaction (RT-PCR) and plaque assays as described elsewhere
[11], and antiaEbola virus antibodies were measured using
standard enzyme-linked immunosorbent assays (ELISAs) [12].
Standard blood counts were evaluated with a Coulter ACA*T diff
(Beckman Coulter). For analysis with flow cytometry, spleens
were ground into single cell suspensions with the BD Medimachine
tissue grinder. After incubation with Fc Block (BD), cells were
washed and incubated with antibody (CD3 FITC BD no. 555274, CD8
V450 BD no. 560469, CD14 PerCP eBio no. 45-0141, CD4 PE eBio no.
12-0041-82, CD11b APC BD no. 553312, and CD19 PE-Cy7 BD no.
557655). Cells were washed with PBS and fixed in BD cytofix.
Data were immediately acquired with a BD FACSCantoII and
analyzed with FlowJo (version 7). The Bio-Plex Mouse Cytokine
23-Plex Panel assay (Bio-Rad 171-F11241) was used to measure
multiple cytokines, chemokines, and growth factors in serum and
tissue supernatants according to the manufacturer's
instructions. Mice that survived the initial infection were
tested for Ebola-specific serological response on day 21 and
rechallenged with the same virus dose without further treatment,
and antibody titers were retested 28 days later.
**RESULTS**
We previously found that rhMBL bound Ebola (Zaire) and Marburg
(Musoke) envelope GPs [2]. RhMBL effectively blocked Ebola GP
interactions with DC-SIGN, and HIV particles lacking gp120/gp41
pseudotyped with Ebola or Marburg GPs were neutralized by the
lectin complement pathway [2]. To develop an in vivo test of
rhMBL effectiveness, we determined that 100 ng/mL of rhMBL was
the minimum concentration needed to inhibit =90% infectivity of
HepG2 cells using Ebola GP pseudotyped lentiviral particles and
to inhibit =90% infectivity of Vero E6 cells using recombinant
Ebola Zaire virus (Mayinga strain)-eGFP (data not shown). We had
previously found that a single intraperitoneal dose of 75 Aug of
rhMBL reconstituted the lectin complement pathway in
MBL-knockout mice [1]. We compared the pharmacokinetic
parameters (Table 1) of that single reconstitution dose (4.3
mg/kg) with a higher single intraperitoneal dose of 350 Aug (20
mg/kg) to identify a potentially supraphysiological dose to test
in model infections. The average maximum serum concentration
(Cmax) of both doses exceeded the minimum concentration of MBL
that inhibited infection in vitro by at least 55-fold. The
average ratio of maximum to baseline complement component 4
cleavage activity was 1.7 for the 75-Aug rhMBL dose and 5.4 for
the 350-Aug dose.
**Table 1.****Pharmacokinetic Parameters of Low- vs High-Dose
Recombinant Human Mannose-Binding Lectin (rhMBL) Therapy in
Uninfected Mice**
Intraperitoneal administration of 100 pfu of native Ebola Zaire
virus (3000 A LD50) is uniformly fatal in mice. Treatment with
75 Aug of rhMBL per dose every 12 hours failed to protect mice
from that virus inoculum. Therefore, we increased rhMBL to 350
Aug administered every 12 hours for 10 days starting either 1
hour before or 12 hours after Ebola virus challenge (Figure 1A
and 1B). When treatment was started 1 hour before virus
infection, the supraphysiological dose increased survival to
> 40% of mice in several trials (Figure 1A). We then started
treatment 12 hours after viral infection. We compared survival
in wild-type and complement component 3 (C3)adeficient mice as
the inhibitory effects of MBL on Ebola virus are mediated by
complement in cell culture [2]. Once again we saw an increase in
survival from 0% to >40% in rhMBL-treated mice, and survival
was dependent on an intact complement pathway, since
C3-deficient mice did not survive (Figure 1B). All inoculated
mice showed signs of infection according to standardized
observation scores and weight loss, and surviving mice had
detectable Ebola virusaspecific antibodies 28 days after
infection (data not shown).
We monitored the effect of treatment started 12 hours after
infection on a variety of laboratory indices. Mean white blood
cell counts were 9100 cells/mL in MBL-treated mice (n = 5)
compared with 4525 cells/mL on day 7 after infection in the
surviving sham-treated mice (n = 4). Average lymphocyte counts
were also higher in MBL-treated mice compared with controls
(5500 cells/mL vs 2800 cells/mL, respectively). A similar trend
was seen for platelet counts, which averaged 726,000 cells/mL in
the treatment group and 239,000 cells/mL in the controls. These
differences were statistically significant for platelet counts
on day 5 (672,000 cells/mL vs 322,000 cells/mL, P = .014; Figure
1C).
In a separate experiment, spleens were harvested on day 5 after
infection (4 sham-treated and 4 MBL-treated mice). Constituent
cell populations were assayed by flow cytometry. Numbers of
splenic CD3-CD19+ cells (B lymphocytes) and CD11b+ granulocytes
were higher in MBL-treated mice (89.2% vs 85.1%, P = .019; 17.6%
vs 12.8%, P = .04, respectively). The RNA viral loads as
determined by RT-PCR in blood, liver, and spleen 5 days after
infection were similar in sham- and rhMBL-treated mice (P >
.05). Virus titers in blood were generally lower on days 1 and 3
in rhMBL-treated mice as determined by plaque assays (P >
.05; Figure 1D). Of 23 cytokines and chemokines tested in serum,
liver, and spleen on day 5 after inoculation, lower values
(fluorescence intensity units) for interleukin (IL)-1b (170 vs
253, P = .07), IL-5 (89 vs 112, P = .03), IL-10 (379 vs 518, P =
.004), IL-13 (264 vs 384, P = .008), and IL-17 (120 vs 174, P =
.028) were found in liver homogenates from rhMBL-treated mice
(Figure 1E). We tested protective immunity in 5 seropositive
mice that survived initial infection by rechallenging them with
native Ebola virus 28 days after initial infection. It is
noteworthy that all MBL-treated survivors also survived the
second viral challenge. Similar or higher immunoglobulin G, A,
and M antibody titers were seen 28 days after the second
challenge with the virus (Figure 1F).
**DISCUSSION**
In the past 3 decades, approved antivirals have increased from a
few nucleoside analogues to well over 40 drugs [13]. The human
immunodeficiency virus (HIV) and hepatitis C virus (HCV)
epidemics particularly drove antiviral discovery toward
rationally designed drugs targeting specific viral enzymes.
Although this approach was remarkably effective, the advent of
newly emerging or drug-resistant viruses that threaten humans
calls for the development of more broadly active agents
targeting viral components shared among viruses. N-glycosylation
of viral envelopes is an important such target shared between
influenza, HIV, HCV, West Nile virus, SARS-CoV, Hendra virus,
Nipah virus, and filoviruses (Ebola and Marburg viruses) [7]. To
assess one possible strategy against N-glycosylated viruses, we
tested a stringent Ebola virus infection model (3000 A LD50) in
mice.
Filovirus infections are characterized by marked lymphopenia,
severe degeneration of lymphoid tissues, dysregulated dendritic
cell function, and cytokine stormsaall hallmarks of pathogens
that subvert both innate and adaptive immune responses [14].
Nevertheless, survivors exhibit detectable virus-specific
antibody responses [15]. Therefore, we hypothesized that
administration of a recombinant innate immune molecule that
targets glycosylated viruses might bridge an infected individual
to recovery. Here we show for the first time that rhMBL can be
used as a therapeutic agent to achieve serum concentrations in
mice that correspond to levels in humans that are 7a24-fold
higher than average human concentrations and complement cleaving
activity that is >5-fold higher than baseline values in mice.
This result confirms our previous in vitro data showing that MBL
possesses complement-dependent intrinsic antimicrobial activity
[2].
Biological responses of the infected mice to rhMBL treatment
further indicated that our strategy targeted the main pathogenic
effects of Ebola viruses. MBL-treated mice had higher B
lymphocyte and CD11b+ granulocyte counts and demonstrated
down-regulation of intrahepatic proinflammatory (IL-1b and
IL-17) and Th2 cytokines (IL-5, IL-10, and IL-13) early in the
course of infection (Figure 1E), suggesting that rhMBL may
mitigate the detrimental effects of the characteristic cytokine
storm. MBL-treated mice tended to have greater inhibition of
viral replication on days 1 and 3 after infection (P > .05;
Figure 1D). Most important, rhMBL treatment bridged surviving
mice to development of an effective adaptive immune response
(Figure 1F). Future experiments will be needed to scale
high-dose rhMBL therapy for use in larger animal models and to
test rhMBL in combination with other promising experimental
therapies such as small molecule inhibitors, coagulation
modulators, antisense technologies, therapeutic antibodies and
cytokines, and postexposure vaccination. In summary, we report
that supraphysiologic rhMBL therapy may be an effective
immunotherapeutic strategy against Ebola virus, and since Ebola
glycoproteins potentially model other glycosylated viruses,
rhMBL therapy may offer a novel broad-spectrum antiviral
approach.
**Funding**
This study was supported by grant U01-AI070330 to E.V.S. from
the National Institutes of Health (NIH). E.V.S. was additionally
supported by NIH grant RO1 CA112021. G.G.O. was additionally
supported by the Defense Threat Reduction Agency Medical
Biological Defense Research Program, Therapeutic Research
Program 4.10007\_08\_RD\_B. K.T. received additional support from
NIH grants 1UO1 AI074503 and 1R21 AI077081.
**Acknowledgments**
Potential conflicts of interest: We declare no commercial
interests that might pose a conflict of interest.
The authors thank Enzon Pharmaceuticals, Bridgewater, New
Jersey, for providing recombinant human mannose-binding lectin,
and members of the Program of Developmental Immunology at
Massachusetts General Hospital for insightful comments.
**References**
1. Shi L, Takahashi K, Dundee J, et al. Mannose-binding
lectin-deficient mice are susceptible to infection with
Staphylococcus aureus. J Exp Med. 2004;199:1379a90. [PMC free
article] [PubMed]
2. Ji X, Olinger GG, Aris S, Chen Y, Gewurz H, Spear GT.
Mannose-binding lectin binds to Ebola and Marburg envelope
glycoproteins, resulting in blocking of virus interaction with
DC-SIGN and complement-mediated virus neutralization. J Gen
Virol. 2005;86:2535a42. [PubMed]
3. Petersen KA, Matthiesen F, Agger T, et al. Phase I safety,
tolerability, pharmacokinetic study of recombinant human
mannan-binding lectin. J Clin Immunol. 2006;26:465a75. [PubMed]
4. Valdimarsson H, Vikingsdottir T, Bang P, et al. Human
plasma-derived mannose-binding lectin: a phase I safety
pharmacokinetic study. Scand J Immunol. 2004;59:97a102. [PubMed]
5. Bang P, Laursen I, Thornberg K, et al. The pharmacokinetic
profile of plasma-derived mannan-binding lectin in healthy adult
volunteers patients with Staphylococcus aureus septicaemia.
Scand J Infect Dis. 2008;40:44a8. [PubMed]
6. Brouwer N, Frakking FN, van de Wetering MD, et al.
Mannose-binding lectin (MBL) substitution: recovery of opsonic
function in vivo lags behind MBL serum levels. J Immunol.
2009;183:3496a504. [PubMed]
7. Vigerust DJ, Shepherd VL. Virus glycosylation: role in
virulence immune interactions. Trends Microbiol. 2007;15:211a8.
[PubMed]
8. Vorup-Jensen T, Sorensen ES, Jensen UB, et al. Recombinant
expression of human mannan-binding lectin. Int Immunopharmacol.
2001;1:677a87. [PubMed]
9. Michelow IC, Dong M, Mungall BA, et al. A novel
l-ficolin/mannose-binding lectin chimeric molecule with enhanced
activity against Ebola virus. J Biol Chem. 2010;285:24729a39.
[PMC free article] [PubMed]
10. Bray M, Davis K, Geisbert T, Schmaljohn C, Huggins J. A
mouse model for evaluation of prophylaxis and therapy of Ebola
hemorrhagic fever. J Infect Dis. 1998;178:651a61. [PubMed]
11. Weidmann M, Muhlberger E, Hufert FT. Rapid detection
protocol for filoviruses. J Clin Virol. 2004;30:94a9. [PubMed]
12. Warfield KL, Posten NA, Swenson DL, et al. Filovirus-like
particles produced in insect cells: immunogenicity protection in
rodents. J Infect Dis. 2007;196:S421aS429. [PubMed]
13. Clercq ED. Three decades of antiviral drugs. Nat Rev Drug
Discov. 2007;6:941.
14. Zampieri CA, Sullivan NJ, Nabel GJ. Immunopathology of
highly virulent pathogens: insights from Ebola virus. Nat
Immunol. 2007;8:1159a64. [PubMed]
15. Baize S, Leroy EM, Georges-Courbot MC, et al. Defective
humoral responses extensive intravascular apoptosis are
associated with fatal outcome in Ebola virus-infected patients.
Nat Med. 1999;5:423a6. [PubMed]
Mannose-binding lectin binds to Ebola and Marburg envelope
glycoproteins, resulting in blocking of virus interaction with
DC-SIGN and complement-mediated virus neutralization.[J Gen
Virol. 2005]
A novel L-ficolin/mannose-binding lectin chimeric molecule
with enhanced activity against Ebola virus.[J Biol Chem. 2010]
Cyanovirin-N binds to the viral surface glycoprotein,
GP1,2 and inhibits infectivity of Ebola virus.[Antiviral Res.
2003]
Ebola virus: unravelling pathogenesis to combat a deadly
disease.[Trends Mol Med. 2006]
Drug targets in infections with Ebola and Marburg
viruses.[Infect Disord Drug Targets. 2009]
Imino sugar glucosidase inhibitors as broadly active
anti-filovirus agents[Emerging Microbes & Infections. 2013]
Activity of and Effect of Subcutaneous Treatment with the
Broad-Spectrum Antiviral Lectin Griffithsin in Two Laboratory
Rodent Models[Antimicrobial Agents and Chemotherapy. 2014...]
Animal models for Ebola and Marburg virus
infections[Frontiers in Microbiology. ]
Lectin-Dependent Enhancement of Ebola Virus Infection via
Soluble and Transmembrane C-type Lectin Receptors[PLoS ONE. ]
A Syrian Golden Hamster Model Recapitulating Ebola
Hemorrhagic Fever[The Journal of Infectious Diseases. 2013]
Recombinant expression of human mannan-binding lectin.[Int
Immunopharmacol. 2001]
A novel L-ficolin/mannose-binding lectin chimeric molecule
with enhanced activity against Ebola virus.[J Biol Chem. 2010]
A mouse model for evaluation of prophylaxis and therapy of
Ebola hemorrhagic fever.[J Infect Dis. 1998]
Rapid detection protocol for filoviruses.[J Clin Virol.
2004]
Filovirus-like particles produced in insect cells:
immunogenicity and protection in rodents.[J Infect Dis. 2007]
Mannose-binding lectin binds to Ebola and Marburg envelope
glycoproteins, resulting in blocking of virus interaction with
DC-SIGN and complement-mediated virus neutralization.[J Gen
Virol. 2005]
Mannose-binding lectin-deficient mice are susceptible to
infection with Staphylococcus aureus.[J Exp Med. 2004]
Mannose-binding lectin binds to Ebola and Marburg envelope
glycoproteins, resulting in blocking of virus interaction with
DC-SIGN and complement-mediated virus neutralization.[J Gen
Virol. 2005]
Review Virus glycosylation: role in virulence and immune
interactions.[Trends Microbiol. 2007]
Review Immunopathology of highly virulent pathogens:
insights from Ebola virus.[Nat Immunol. 2007]
Defective humoral responses and extensive intravascular
apoptosis are associated with fatal outcome in Ebola
virus-infected patients.[Nat Med. 1999]
Mannose-binding lectin binds to Ebola and Marburg envelope
glycoproteins, resulting in blocking of virus interaction with
DC-SIGN and complement-mediated virus neutralization.[J Gen
Virol. 2005]
---
[**http://www.ncbi.nlm.nih.gov/pubmed/15613479**](http://www.ncbi.nlm.nih.gov/pubmed/15613479)**J Biol Chem. 2005 Mar 11;280(10):9345-53. Epub 2004 Dec
21.**
**Isolation and characterization of
griffithsin, a novel HIV-inactivating protein, from the red
alga Griffithsia sp.**
**Mori T, O'Keefe BR, Sowder RC, Bringans S, Gardella R, Berg
S, Cochran P, Turpin JA, Buckheit RW Jr, McMahon JB, Boyd
MR.**
**Abstract**
Griffithsin (GRFT), a novel anti-HIV protein, was isolated from
an aqueous extract of the red alga Griffithsia sp. The 121-amino
acid sequence of GRFT has been determined, and biologically
active GRFT was subsequently produced by expression of a
corresponding DNA sequence in Escherichia coli. Both native and
recombinant GRFT displayed potent antiviral activity against
laboratory strains and primary isolates of T- and M- tropic
HIV-1 with EC50 values ranging from 0.043 to 0.63 nM. GRFT also
aborted cell-to-cell fusion and transmission of HIV-1 infection
at similar concentrations. High concentrations (e.g. 783 nM) of
GRFT were not lethal to any tested host cell types. GRFT blocked
CD4-dependent glycoprotein (gp) 120 binding to
receptor-expressing cells and bound to viral coat glycoproteins
(gp120, gp41, and gp160) in a glycosylation-dependent manner.
GRFT preferentially inhibited gp120 binding of the monoclonal
antibody (mAb) 2G12, which recognizes a carbohydrate-dependent
motif, and the (mAb) 48d, which binds to CD4-induced epitope. In
addition, GRFT moderately interfered with the binding of gp120
to sCD4. Further data showed that the binding of GRFT to soluble
gp120 was inhibited by the monosaccharides glucose, mannose, and
N-acetylglucosamine but not by galactose, xylose, fucose,
N-acetylgalactosamine, or sialic acid-containing glycoproteins.
Taken together these data suggest that GRFT is a new type of
lectin that binds to various viral glycoproteins in a
monosaccharide-dependent manner. GRFT could be a potential
candidate microbicide to prevent the sexual transmission of HIV
and AIDS.
---
**US2010331240**
**METHODS FOR PREVENTION AND TREATMENT OF INFECTIONS WITH
SUPRAPHYSIOLOGICAL DOSES OF MANNAN-BINDING LECTIN (MBL)
AND FICOLIN-MBL FUSION PROTEINS**
Inventor: MICHELOW IAN // SCHMIDT EMMETT
The present invention provides methods of treatment and/or
prevention of infections, for example, viral and bacterial
infections, in individuals, wherein the method comprises
administering a supraphysiological amount of mannose-binding
lectin (MLB) and/or ficolin-MBL fusion protein to an individual
afflicted with an infection or at risk of an infection, such as
a bacterial or a viral infection. For example, methods for
treatment and/or prevention of Ebola virus infection are
provided.
**BACKGROUND OF THE INVENTION****[0002] 1. Field of the Invention**
[0003] The present invention pertains to the use of subunits and
oligomers of mannan-binding lectin (MBL) and ficolin-MBL fusion
proteins for prevention and/or treatment of infections,
particularly in subjects who have normal and functional MBL
serum levels.
**[0004] 2. Background of the Invention**
[0005] Infections count for a large part of morbidity and
mortality in the world. While bacterial infections have been
tackled by antibiotics and bacteriophages, new treatment methods
are sorely needed for the growing amount of bacteria that have
become resistant to these treatments. Viruses are a difficult
target for treatment in humans and other animals because they
use animal cells to replicate and spread. While some viral
infections can be prevented using vaccination or antibody-based
therapies, several serious and lethal viruses remain currently
without effective treatment.
[0006] One of such lethal virus family is filoviruses. The two
most known lethal filoviruses are Ebola and Marburg viruses.
Ebola and Marburg virus can cause acute, lethal hemorrhagic
fevers for which no vaccines or effective treatments currently
exist. Marburg and Ebola envelope glycoproteins consist of
glycoprotein 1 (GP1) and membrane-bound glycoprotein 2 (GP2)
protein that are covalently linked by a disulfide bond (Sanchez
et al., Proc Natl Acad Sci USA 93:3602-3607, 1996). Although the
causes of filovirus virulence are not well known, there is
evidence that glycans on the viral glycoproteins play distinct
roles in pathogenesis of these viruses (Takeda and Kawaoke,
Trends Microbiol 9:506-511, 2001).
[0007] It would be useful to discover and develop new treatments
for infections, such as viral and bacterial infections that
could be used in prevention and/or treatment of infections
and/or to supplement the currently available treatment methods
to combat infections. In addition, it would be useful to
discover new treatments for infectious diseases that do not
currently have an effective treatment method, such as filovirus
infection or infections by bacteria that have developed
resistance to the available antibiotics.
**SUMMARY OF THE INVENTION**
[0008] The present invention is directed to methods of treatment
and/or prevention of infections, for example, viral and
bacterial infections, in individuals, wherein the method
comprises administering a supraphysiological amount of
mannose-binding lectin (MLB) or ficolin-MBL fusion protein to an
individual afflicted with an infection or at risk of infection,
such as a viral or bacterial infection.
[0009] The invention is based upon a surprising discovery, that
an infection in an individual with normal MBL serum
concentration and function, i.e., who has no defect in MBL, can
be successfully treated or prevented by using supraphysiological
amounts of MBL or by using ficolin-MBL fusion protein.
[0010] The terms "supraphysiological" or "supraphysiologic" are
intended to encompass amounts of MBL or ficolin-MBL fusion
protein that exceed the normal serum concentration of MBL in an
individual, preferably a human individual. The normal serum
concentration of MBL can be either measured individually, or
estimated based upon a normal range or average normal serum
concentration in humans or particular human populations.
Typically, the "normal" human serum concentration of MBL is
considered a concentration in individuals who do not carry
genetic alterations or mutations that are known to reduce the
amount or function of MBL in said individual.
[0011] In one embodiment, and all other embodiments described
herein, one uses amounts of MBL that result in blood
concentration of >2\* to 10\* the average human serum
concentration, which is considered a normal serum concentration.
In one embodiment, the human average MBL serum concentration is
estimated to be about 2 [mu]g/mL. Accordingly, one can use any
amount that results in serum concentration of between 4-20
[mu]g/mL. For example, an amount that results in serum
concentration of about 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,
16, 17, 18, 19, or 20 [mu]g/mL In one embodiment, similar
amounts of ficolin-MBL are used.
[0012] Viral infections that can be prevented,
ameliorated/treated or cured using the methods of the present
invention include, but are not limited to, filoviruses,
including Ebola and Marburg viruses, HIV, influenza, severe
acute respiratory syndrome coronavirus (SARS-CoV), hepatitis B
virus, hepatitis C virus, respiratory syncytial virus, and
herpes simplex virus.
[0013] Bacterial infections that can be prevented
ameliorated/treated or cured using the methods of the present
invention include, but are not limited to Staphylococcus aureus;
Neisseria meningitidis; Burkholderia multivorans, group B.
streptococcus, Escherichia coli, Pseudomonas aeruginosa,
Mycoplasma pneumoniae, and Chlamydia pneumoniae.
[0014] While the methods of the invention can be used for
treatment and/or prevention of infections in any animal or bird,
a preferred target individual is human.
[0015] In one embodiment, and all other embodiments described
herein, the target individual is affected with a bacterium that
has become resistant to currently available antibiotics. In one
embodiment, one uses the method of the present invention in
combination with antibiotics or bacteriophages, or anti-viral
agents.
[0016] In one embodiment, and all other embodiments described
herein, the individual is affected with a filovirus, such as
Ebola or Marburg virus.
[0017] In one embodiment, and all other embodiments described
herein, the method comprises first selecting a patient who is
infected with a virus or bacterium, and then administering to
the selected individual a supraphysiological amount of MLB or
ficolin-MBL fusion protein.
[0018] In one embodiment, and all other embodiments described
herein, the individual affected with, exposed to or susceptible
to be exposed to an infection, such as bacterial or viral
infection does not have a congenital or acquired MBL deficiency.
In one embodiment, and all other embodiments described herein,
one first determines if the individual has a congenital or
acquired MBL deficiency. If the individual does not have such a
deficiency, the individual can be administered a
supraphysiological amount of MBL or ficolin-MBL fusion protein
as a treatment or preventive measure to fight a viral infection
or a suspected viral infection or exposure to an environment
likely to carry viruses, such as an Ebola virus.
[0019] MBL can be purified from natural sources or from material
produced by recombinant technologies, or by any other suitable
MBL-producing cell line, for the prophylaxis and/or treatment of
infections. Preparations and pharmaceutical compositions of MBL
are known. In one embodiment, one uses the MBL as described in
U.S. Pat. No. 5,270,199, which is herein incorporated by
reference in its entirety. Also preparations and pharmaceutical
compositions of ficolin-MBL fusion proteins are known. In one
embodiment, one uses ficolin-MBL chimeric proteins described in,
e.g., U.S. Patent Application Publication No. 20060188963. In
one embodiment, one uses SEQ ID NO: 1 to produce MBL. Ficolin
sequences, for example SEQ ID NO: 3, and SEQ ID NO: 8 can be
used to make constructs for recombinantly producing various
ficolin-MBL fusion proteins.
[0020] One aspect of the invention relates to treatment and/or
prophylaxis of infections in individuals affected with a viral
or bacterial infection using supraphysiological amount of MBL or
ficolin-MBL fusion protein. In one embodiment, the individuals
are not immunocompromised.
[0021] Without wishing to be bound by a theory, we believe that
MBL exerts its antimicrobial activity mainly through its
opsonizing activity (preparation of microorganisms for
phagocytosis). This activity is dependent on activation of
complement after binding of MBL to the microbial surface and
deposition of C4b and C3b on the microorganism. MBL can also
promote direct complement-mediated killing of the microorganism
through an activation of the terminal lytic pathway of
complement and insertion of the membrane attack complex (MAC) in
the membrane. Without wishing to be bound by a theory, this
mechanism is considered of minor importance. Many
microorganisms, such as Gram-positive bacteria, e.g.,
Streptococcus pneumonia, are resistant to MAC, but can be
eliminated by opsonophagocytosis. The inhibition of infection
may be mediated by MBL directly neutralizing the pathogen,
enhancing uptake by phagocytic cells that eliminate the
infection, or by killing the pathogens by activation of the
complement protein pathway.
[0022] Because the MBL is normally present at physiological
amounts in individuals who do not have congenital defects in it
or who are not immunocompromised, it was surprising that one can
exert a virus dose reducing effect by administering additional,
supraphysiological amount of MBL into such an individual.
[0023] In another aspect, the present invention relates to the
use of a composition comprising at least one mannan-binding
lectin (MBL) subunit, or at least one oligomer comprising the at
least one mannan-binding lectin (MBL) subunit, in the
manufacture of a medicament for prophylactic, ameliorating or
curative treatment of an infection, including a viral or
bacterial infection, in an individual initially having plasma
levels of MBL of about 5 [mu]g/mL. In one embodiment, the
individual is not genetically disposed to an MBL deficiency or
does not have acquired MBL deficiency.
[0024] Accordingly, in one embodiment, the methods are used as
prophylaxis for individuals who are likely to be exposed, or who
have already been exposed to viruses and/or bacteria, but do not
yet have symptoms of infection, wherein the presence of
supraphysiological amount of MBL or ficolin-MBL will prevent
infection or ameliorate symptoms of an infection.
[0025] In one embodiment, the invention provides a method of
preventing a filovirus infection by administering a
supraphysiological dose of MBL or ficolin-MBL fusion protein to
an individual who is likely to be exposed to a filovirus. In one
embodiment, the filovirus is Ebola virus. In one embodiment, the
filovirus is Marburg virus.
[0026] In one embodiment, the invention provides use of MBL or
ficolin-MBL fusion protein as a medicament for treatment of
infections, particularly viral and bacterial infections, in
amounts that are supraphysiological.
**BRIEF DESCRIPTION OF DRAWINGS****[0027] FIG. 1 shows a schematic drawing of the
mannose-binding lectin (MBL) protein and L-ficolin.****[0028] FIG. 2 shows a schematic drawing showing the
functional and structural domains of MBL and L-ficolin.****[0029] FIG. 3 shows a schematic drawing showing the
construction of the three chimeric FCN-MBL fusion proteins.****[0030] FIG. 4 shows an SDS-PAGE protein gel showing the
purified recombinant chimeric FCN-MBL fusion proteins and the
denatured purified recombinant MBL under reducing conditions.****[0031] FIG. 5 shows a protein gel of the purified
recombinant chimeric FCN-MBL fusion proteins and recombinant
MBL under non-reducing conditions.****[0032] FIG. 6 shows a competitive ELISA comparing avidity
of rhMBL and chimeric proteins (100 ng each) binding to
mannan.****[0033] FIG. 7 shows a C4 deposition assay. The C4
deposition assay is an ELISA-based functional assay that
measures the relative capacities of MBL or the chimeric
protein to bind human C4. Mannan (10 ug/mL) is coated on a
96-well ELISA plate, blocked with BSA, and incubated with
varying concentrations of rhMBL or chimeric proteins. Human C4
(10 ug/mL) is then added and detected with biotin-streptavidin
conjugated antibodies. FCN-MBL76 had significantly greater C4
binding activity compared with rhMBL and the other chimerics.
This result suggests that FCN-MBL76 has greater complement
pathway activating capacity which may result in enhanced
pathogen lysis or neutralization.****[0034] FIG. 8 shows calreticulin binding assay. The
96-well ELISA plate was coated with rhMBL or chimeric proteins
(10 ug/mL), blocked with BSA and incubated with 5 ug/mL
biotinylated human placental calreticulin that was measured at
absorbance O.D. 405. FCN-MBL76 bound to human placental
calreticulin significantly better than rhMBL or the other
chimeric proteins. This may have important implications for
the relative functions of the proteins because calreticulin is
the putative cellular receptor on phagocytes for native MBL
and therefore, enhanced binding of the chimeric molecule may
result in improved pathogen clearance by opsonophagocytosis.****[0035] FIG. 9 shows an inhibition assay using Hep G2
cells infected with lentivirus (HIV) pseudotyped with Ebola
glycoprotein. Hep G2 cells at approximately 80% confluence in
96-well tissue culture plates were infected with HIV particles
without an envelope (HIV-env neg; solid square) or with an
envelope consisting of Ebola glycoprotein (other symbols). The
virions encoded luciferase that was expressed only in infected
cells and detected with a commercial luciferase assay. Before
addition of viral particles to the cells, the viruses were
preincubated with 0, 0.1 or 1 ug/mL of rhMBL or chimeric
proteins in veronal-buffered saline with 5 mM CaCl2 for 1 hour
at 37 C. Infection was achieved by spinoculation of cells at
1000 g\*2 hrs. The viral protein mixture was replaced with EMEM
culture media and incubated at 37 C for 40 hrs after which,
the cells were lyzed and luciferase expression was quantified.
rhMBL and the chimeric proteins inhibited viral infection to
similar significant extents (1 ug/mL vs no protein,
p<0.001)****[0036] FIG. 10 shows an inhibition assay using Hep G2
cells infected with native Ebola-Zaire virus. 30,000 Hep G2
cells/well in 96-well tissue culture plates were infected with
native Ebola virus (Zaire strain) that was genetically
engineered to express GFP. The viral particles were
preincubated with 0, 0.1 or 1 ug/mL of rhMBL or chimeric
proteins in veronal-buffered saline with 10 mM CaCl2 for 1
hour at 37 C. The viral protein mixture was added to the cells
and incubated for 48 hrs after which time the cells were
washed. Viral infection of cells was quantified by measuring
GFP expression. rhMBL and the chimeric proteins inhibited
viral infection but FCN-MBL76 was the most effective.****[0037] FIG. 11 shows that a pharmacokinetic modeling of
rhMBL (recombinant human MBL) in immunocompetent C57B/6J mice
revealed that doses of 75 mcg and 350 mcg doses produced Cmax
of ~5 [mu]g/mL and ~15 [mu]g/mL, respectively and half-life of
~11 hours at both doses. A previous study showed that 75 [mu]g
is the minimum dose of rhMBL required to activate complement
in an MBL-deficient mouse model.****[0038] FIG. 12 shows a Kaplan Meier survival analyses:
350 [mu]g rhMBL was given immediately pre-challenge with EBOV
Zaire and continued every 12 hrs\*10 days resulting in 42%
survival rate (log rank, p<0.008).****[0039] FIG. 13 shows a Kaplan Meier survival curve with a
post-challenge analysis which demonstrated that recombinant
human MBL-treated wild-type mice had a significant survival
advantage: 40% survived compared to 100% mortality among
wild-type and C3 knock-out mice treated with saline or rhMBL
indicating that rhMBL provides protection but that the
protection is dependent on C3. MBL treated mice survived
significantly longer than mice not treated with MBL. EBOV was
administered IV 100pfu (plaque forming units) 3000xLD50. WT
(wildtype, C57B/6J mice) versus C3 knock out (KO). Recombinant
MBL (rhMBL) was administered at 350 mcg IL 12 hors post
challenge, then q12hx10 days vs. sham Rx.\*log rank,
p<0.0004.****[0040] FIGS. 14A-14D show that sham treated wild-type
mice all died before the 10 day time point. rhMBL-treated
wild-type mice had significantly higher total white blood cell
and lymphocyte counts after day 5 suggesting that lymphocyte
responses in these mice may be protective.****[0041] FIG. 15 shows A Rush HepG2 Infection Assay for
HIV-EBOZ vs. HIV-env as a negative control with 400 pg/well,
96 well format. MDS (M.R. 1:2, non-HI). The results
demonstrate that MBL significantly inhibited infection of
HepG2 cells by HIV particles pseudotyped with Ebola
glycoprotein. The control virus is an HIV particle without
viral surface glycoproteins.** **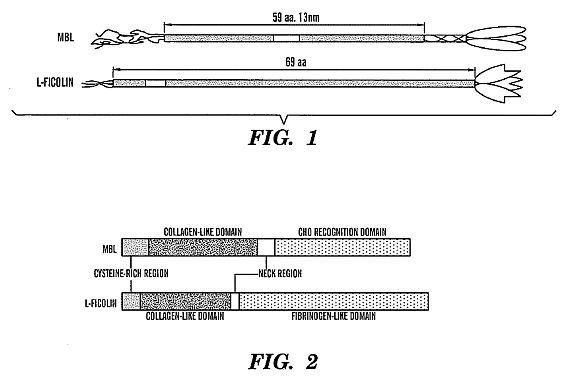 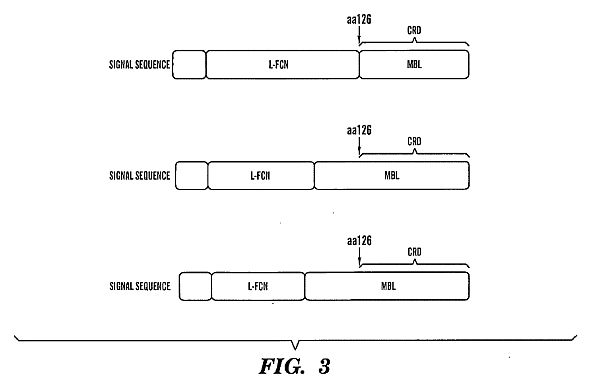 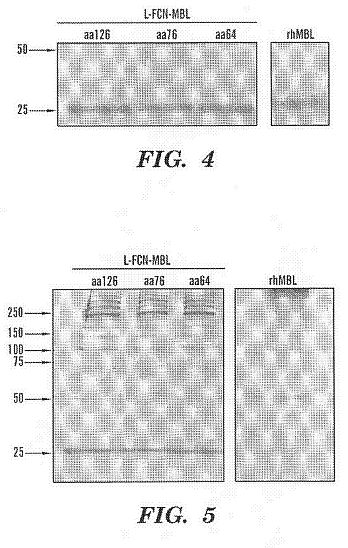 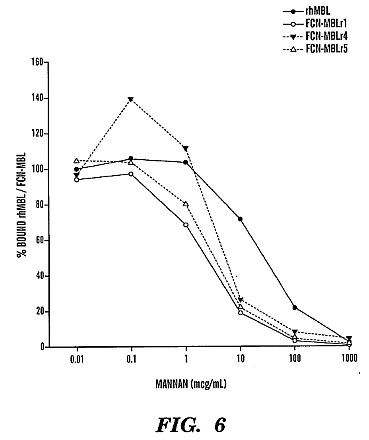 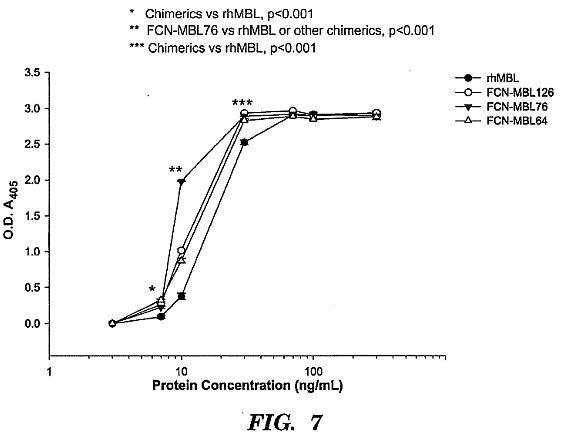 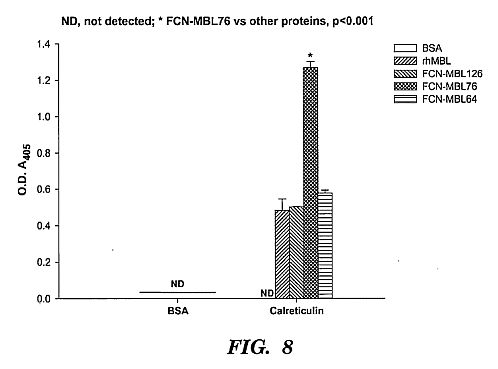 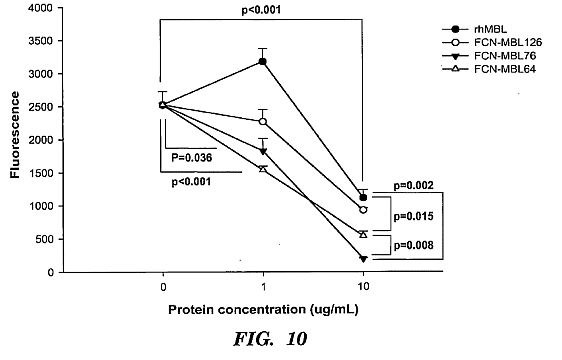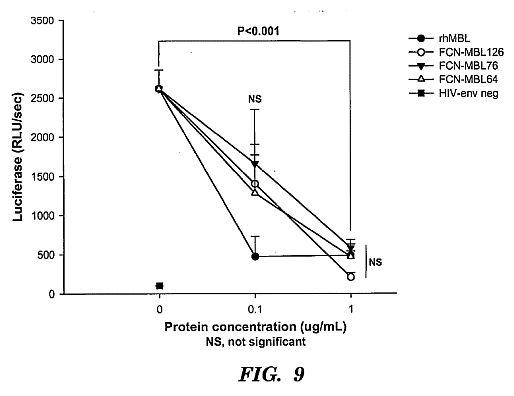 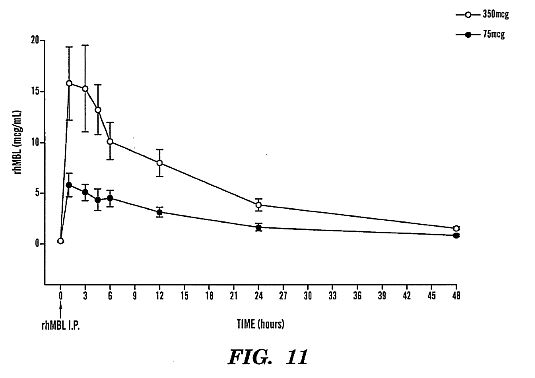 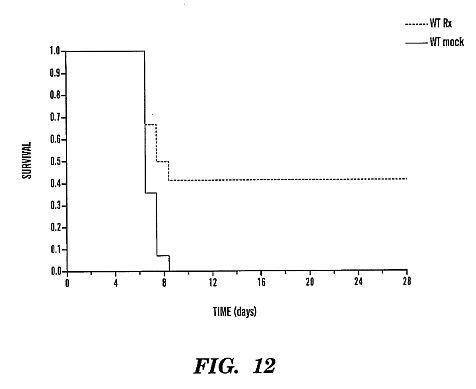 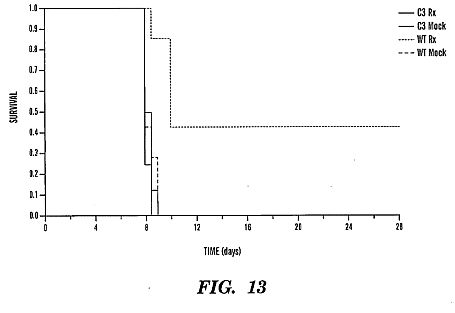 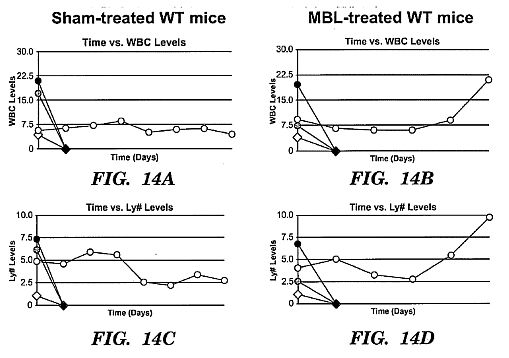 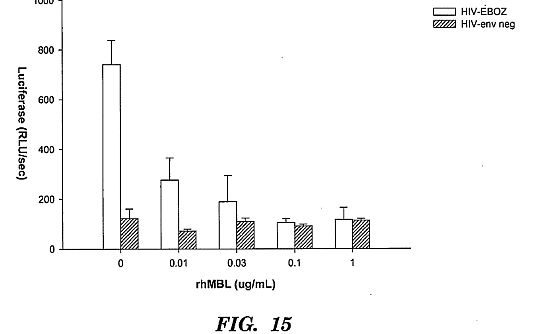****DETAILED DESCRIPTION OF THE INVENTION**
[0042] The present invention is directed to methods and uses of
MBL and ficolin-MBL fusion proteins for the treatment of
infections.
[0043] The innate immune system that defends humans from
infections is comprised of a network of recognition and effector
molecules that act together to protect the host in the first
minutes or hours of exposure to an infectious challenge.
[0044] The mannan-binding lectin (MBL), synonymous to
mannose-binding lectin, mannan-binding protein or
mannose-binding protein (MBP), is an evolutionarily conserved
circulating host defense protein that acts as a broad spectrum
recognition molecule against a wide variety of infectious agents
(see, e.g., review by Takahashi et al. Current Opinion in
Immunology 18:16-23, 2006).
[0045] Several groups of lectins, i.e., carbohydrate-binding
proteins, are known in humans. One group is the C-type lectins.
The C-type lectins contain a calcium-dependent carbohydrate
recognition domain (a C-type CRD)(Weis W I, et al. Immunological
Reviews 163: 19-34, 1998). MBL belongs to the subgroup of C-type
lectins, termed collectins, since these soluble proteins are
composed of subunits presenting three CRDs attached to a
collagenous stalk (Holmskov, U., et al., Immunol. Today
15:67-74, 1994). MBL interacts with carbohydrates presented by a
wide range of micro-organisms playing an important role in the
innate immune defense (Turner, M. W. Immunol. Today 17:532-540,
1996 and Takahashi et al., Current Opinion in Immunology,
18:16-23, 2006). When bound to carbohydrate MBL is able to
activate the complement system.
[0046] The complement system may be activated via three
different pathways: the classical pathway, the alternative
pathway, and the third pathway, the mannan-binding lectin (MBL)
pathway, which is initiated by the binding of MBL to
carbohydrates presented by micro-organisms. The components of
the alternative pathway and of the MBL pathway are parts of the
innate immune defense, also termed the natural or the
non-clonal, immune defense, while the classical pathway involves
cooperation with antibodies of the specific immune defense
(Janeway C A, Travers P, Walport M and Capra J D, 1999,
Immunobiology, the immune system in health and disease, Fourth
Edition, Churchill Livingstone).
[0047] The human MBL protein is composed of up to 18 identical
32 kDa polypeptide chains (Lu, J., et al., (1990) J. Immunol.
144:2287-2294), each comprising a short N-terminal segment of 21
amino acids including three cysteine residues, followed by 7
repeats of the collagenous motif Gly-X-Y interrupted by a Gln
residues followed by another 12 Gly-X-Y repeats. A small 34
residue 'neck-region' joins the C-terminal
Ca<2+>-dependent lectin domain of 93 amino acids with the
collagenous part of the molecule (Sastry, K., et al., (1989) J.
Exp. Med. 170:1175-1189).
[0048] The collagenous regions of the three polypeptide chains
combine to form a subunit which is stabilized covalently by
disulphide bridges. Individual subunits are joined by disulphide
bridges as well as by non-covalent interactions (Lu, J., et al.,
J. Immunol. 144:2287-2294, 1990).
[0049] The position of these disulphide bridges has, however,
not been fully resolved. SDS-PAGE analysis under non-reducing
conditions of MBL shows bands with an apparent molecular weight
(m.w.) larger than 200 kDa presumably representing blocks of 3,
4, 5 and even 6 assembled subunits (Lu, J., et al., J. Immunol.
144:2287-2294, 1990).
[0050] The actual number of subunits in the natural human MBL
protein has been controversial. Lipscombe et al. (1995) obtained
data by use of ultracentrifugation suggesting 25% of human serum
MBL to be made of 2-3 subunits and only a minor fraction
reaching the size of 6 subunits (Lipscombe, R. J., et al.,
Immunology 85:660-667, 1995). The relative quantification was
carried out by densitometry of Western blots developed by
chemiluminescence (Lu, J., et al., J. Immunol. 144:2287-2294,
1990) found by SDS-PAGE analysis of fractions from ion exchange
chromatography that the predominant species of covalently linked
MBL subunit chains consisted of tetramers while only pentameric
or hexameric complexes activated complement. Gel permeation
chromatography (GPC) analysis, in contrast, suggests that MBL is
comparable in size with the C1 complex. GPC can be carried out
under conditions which allow for a study of the importance of
weak protein-protein interactions in the formation of MBL
molecules. MBL content in the GPC fractions can be determined by
standard MBL assay techniques.
[0051] MBL is synthesized in the liver by hepatocytes and
secreted into the blood. It binds to carbohydrate structures on
bacteria, yeast, parasitic protozoa and viruses, and exhibits
antibacterial activity through killing of the microorganisms by
activation of the terminal, lytic complement components or
through promotion of phagocytosis (opsonization). The sertiform
structure of MBL is quite similar to the bouquet-like structure
of C1q, the immunoglobulin-binding subcomponent of the first
component in the classical pathway (Turner, M. W.
Mannose-binding lectin: the pluripotent molecule of the innate
immune system. Immunol. Today 17:532-540, 1996). C1q is
associated with two serine proteases, C1r and C1s, to form the
C1 complex. Similarly, MBL is associated with two serine
proteases MASP-1 (Matsushita, M. and Fujita, T, J. Exp. Med.
176:1497-1502, 1992) and MASP-2 (Thiel S, et al., Nature,
386(6624): 506-510, 1997), and an additional protein called
Map19 (Stover C M, et al., J Immunol 162: 3481-3490, 1999).
MASP-1 and MASP-2 have modular structures identical to those of
C1r and C1s (Thiel S, et al., Nature, 386(6624): 506-510, 1997).
The binding of MBL to carbohydrates induces the activation of
MASP-1 and MASP-2. MASP-2 then generates the C3 convertase,
C4b2a, through cleavage of C4 and C2. Reports suggest that
MASP-1 may activate C3 directly. Nothing is known about the
stoichiometry and activation sequence of the MBL/MASP complexes.
MBL has also been characterized in other animals such as
rodents, cattle, chicken and monkeys.
[0052] Based on presence and function of MBL in at least
rodents, cattle, chicken and monkeys, in addition to humans,
makes the methods of the present invention applicable to at
least these animals as well.
[0053] Human mannose-binding protein has been disclosed in U.S.
Pat. No. 5,270,199. Moreover, use of MBL in treatment of
immunocompromised individuals has been described (U.S. Pat. Nos.
6,562,784 and 7,202,207, and U.S. Patent Application Publication
No. 2007-0197428). However, because MBL is a naturally occurring
molecule present in the serum, no one has suggested its use in
treatment or prevention of infections in individuals with normal
serum concentration of MBL. Our discovery that
supraphysiological amounts of MBL can increase the infection
fighting capacity of an individual with normal MBL
concentrations and function was thus surprising.
[0054] Accordingly, one aspect of the invention provides a
method for prevention and treatment of infections in
individuals, such as human individuals, comprising administering
to said individual a supraphysiological amount of MBL.
[0055] The term "supraphysiological" as used in the present
application means amounts greater than the physiological amount
normally present in an individual or greater than minimal
concentration of MBL required to activate a complement, i.e. to
bind to C4. Similar concentrations of ficolin-MBL fusion
proteins can also be used.
[0056] In one embodiment, one uses MBL and/or ficolin-MBL fusion
proteins or combinations thereof in the amount that results in
the amount of about 2-10 times greater than the physiological
amount of MBL in an individual. In one embodiment, one uses, for
example, 2, 3, 4, 5, 6, 7, 8, 9 or 10 times greater than the
physiological amount in the individual. In one embodiment, the
average physiological amount is considered about 2 [mu]g/mL. In
one embodiment, one first determines the physiological amount of
MBL in an individual prior to administering MBL or ficolin-MBL
fusion protein composition to said individual. This is
particularly useful when using MBL or ficolin-MBL as a
prophylaxis for individuals who will be at risk of encountering
infective agents, such as medical personnel or armed forces who
may be a target for a biological attack.
[0057] The concentration of MBL in human serum is largely
genetically determined, but reportedly increases up to threefold
during acute phase infection reactions (Thiel S, et al., Clin
Exp Immunol 90: 31-35, 1992). Three mutations causing structural
alterations and two mutations in the promotor region are
associated with MBL deficiency (Madsen, H. O., et al.,
Immunogenetics 40:37-44, 1994). MBL deficiency is associated
with susceptibility to a variety of infections (Summerfield J A,
et al., Lancet 345: 886-889, 1995; Garred P, et al., Lancet 346:
941-943, 1995).
[0058] It has been estimated that the average physiological
amount of MBL in human serum is about 2 [mu]g/mL. Accordingly,
in one embodiment, one uses the average physiological amount as
the physiological amount, and consequently, the
supraphysiological amount of MBL or ficolin-MBL according to the
present invention is about 2-10 times above the average
physiological MBL level.
[0059] In one embodiment, one takes into account the impact of
MBL haplotypes when considering the physiological serum MBL
concentrations. At least ten distinct MBL haplotypes have been
described in human, four of which (LYPB, LYQC, HYPD and LXPA)
dictate low serum MBL concentrations (Madsen et al. J Immunol
161:3169-3175, 1998; Takahashi et al., Current Opinion in
Immunology 18:16-23, 2006). Human populations from diverse
geographic locations and ethnic and genetic backgrounds have
higher rate of haplotype variation, with a rate of
heterozygosity from 15% in white populations to 30% in certain
African populations. Accordingly, in one embodiment, to
establish the physiological serum MBL level to adjust the amount
of MBL used in the methods of the present invention, one
correlates the level of MBL with a functional measurement of the
MBL:MASP pathway (Takahashi et al., Current Opinion in
Immunology 18:16-23, 2006; Petersen et al., J Immunol Methods
257:107-116, 2001). Accordingly, if the average serum
concentration of MBL in a particular individual or population is
higher, the supraphysiologic dosage is adjusted accordingly.
Similarly, if the average serum concentration of MBL is lower, a
lower amount is needed for the treatment of prevention of
infections. A skilled artisan is easily able to make these
determinations based on the description herein.
[0060] A wide range of oligosaccharides can bind to MBL. As the
target sugars are not normally exposed on mammalian cell
surfaces at high densities, MBL does not usually recognize
self-determinants, but is well suited to interactions with
microbial cell surfaces presenting repetitive carbohydrate
determinants. In vitro, yeast (Candida albicans and Cryptococcus
neoformans), viruses (HIV-1, HIV-2, HSV-2, and various types of
influenza A) and a number of bacteria have been shown to be
recognized by MBL. In the case of some bacteria, the binding
with MBL is impaired by the presence of a capsule (van Emmerik,
L C, et al., Clin.Exp.Immunol. 97:411-416, 1994). However, even
encapsulated bacteria (Neisseria meningitidis) can show strong
binding of MBL (Jack D L, et al., J Immunol 160: 1346-1353,
1998), and is thus one target infection according to the present
invention.
[0061] The microorganisms, which infect MBL deficient
individuals, represent many different species of bacterial,
viral and fungal origin (Summerfield J A, et al., BioMed J 314:
1229-1232, 1997; Miller, M. E., et al., Lancet: 60-63, 1968;
Super, M., et al., Lancet 2:1236-1239, 1989; and Nielsen, S. L.,
et al., Clin. Exp. Immunol. 100:219-222, 1995). Deficiency is
also associated with habitual abortions (Christiansen, O. B., et
al., Scand. J. Immunol., 49, 193-196, 1999). Indeed, MBL appears
to be a general defense molecule against most bacteria, and thus
be considered as one reason why so many bacteria are
non-pathogenic.
[0062] Accordingly, in one embodiment, the methods of the
invention pertain to prevention and/or treatment of infections
caused by any of the foregoing infective agents, including
viruses, yeast, fungus, and bacteria.
[0063] While accumulating data support the notion of a
protective effect of MBL there are also observations suggesting
that infections with some microorganisms, notably intracellular
pathogens, attain a higher frequency in MBL sufficient than in
MBL deficient individuals (Garred, P, et al., Eur. J. Immunogen.
21:125-131, 1994; Hoal-Van Helden E G, et al., Pediatr Res
45:459-64, 1999). This is in concordance with the results of an
animal experiment, where an increased number of HSV-2 were found
in the liver of mice pre-injected with human MBL (Fischer, P B,
et al., Scand J Immunol 39:439-445, 1994). Our results
contradict these findings by showing a strong protective and
treatment effect of administering to a subject a
supraphysiological amount of MBL and/or ficolin-MBL fusion
proteins or combinations thereof.
[0064] Clinical grade MBL has been obtained from blood donor
plasma and shown to be safe upon infusion (Valdimarsson, H., M.
et al., Scand. J. Immunol. 48:116-123, 1998). Accordingly, one
can use such preparations in the methods of the present
invention. Similarly, one can make recombinant MBL using any
well known gene expression system.
[0065] Ficolins, like MBL, are lectins that contain a
collagen-like domain. However, unlike MBL, they have a
fibrinogen-like domain, which is similar to fibrinogen beta- and
gamma-chains. Ficolin also forms oligomers of structural
subunits, each of which is composed of three identical 35 kDa
polypeptides. Each subunit is composed of an amino-terminal,
cysteine-rich region; a collagen-like domain that consists of
tandem repeats of Gly-Xaa-Yaa triplet sequences (where Xaa and
Yaa represent any amino acid); a neck region; and a
fibrinogen-like domain. The oligomers of ficolins comprise two
or more subunits, especially a tetrameric form of ficolin has
been observed.
[0066] Some of the ficolins trigger an activation of the
complement system substantially in similar way as done by MBL.
This triggering of the complement system results in the
activation of novel serine proteases (MASPs).
[0067] The fibrinogen-like domain of several lectins has a
similar function to the CRD of C-type lectins including MBL, and
function as pattern-recognition receptors to discriminate
pathogens from self.
[0068] Serum ficolins have a common binding specificity for
GlcNAc (N-acetyl-glucosamine), elastin or GalNAc
(N-acetyl-galactosamine). The fibrinogen-like domain is
responsible for the carbohydrate binding. In human serum, two
types of ficolin, known as L-ficolin (also called P35, ficolin
L, ficolin 2 or hucolin) and H-ficolin (also called Hakata
antigen, ficolin 3 or thermolabile b2-macroglycoprotein), have
been identified, and both of them have lectin activity.
L-ficolin recognises GlcNAc and H-ficolin recognises GalNAc.
Another ficolin known as M-ficolin (also called P35-related
protein, ficolin 1 or ficolin A) is not considered to be a serum
protein and is found in leucocytes and in the lungs. L-ficolin
and H-ficolin activate the lectin-complement pathway in
association with MASPs. M-Ficolin, L-ficolin and H-ficolin has
calcium-independent lectin activity.
[0069] Accordingly, in one embodiment, the invention provides
methods of prevention and/or treatment of infectious diseases
using MBL-L-ficolin or MBL-H-ficolin fusion proteins, or a
combination thereof.
[0070] Naturally, one can also use a combination of MBL and
MBL-ficolin, such as MBL-L-ficolin and/or MBL-H-ficolin.
[0071] Chimeric molecules of MBL and ficolin have been
described, for example, in U.S. Patent Application Publication
No. 2006-0188963. Although it has been suggested that the
chimeric molecules could be used to prevent and/or treat
infections in patients having clinical symptoms associated with
congenital or acquired MBL deficiency or being at risk of
developing such symptoms (Id.), no one has proposed or shown
that individuals with normal MBL activity would benefit from
additional, supraphysiological amounts of MBL or MBL-ficolin in
combating infectious diseases.
[0072] Based on our findings, the present invention provides a
novel method for treatment or prevention of infections in an
individual having normal expression and normal function of MBL.
[0073] In addition, fusion proteins useful according to the
methods of the invention can designed in such a way as to test
whether the source of the MASP-binding site and flanking
sequences, and presence of the "kink" from MBL affect ligand
binding activity and/or complement activation. Without wishing
to be bound by a theory, we designed the proteins in the
examples based on the assumption that differences at these sites
alter the protein conformational structure which in turn alters
protein-protein interactions. Therefore any differences in
protein activity can assist in understanding the functional
parts of the molecules. We discovered that FCN-MBL76 has a
greater activity in various assays. Without wishing to be bound
by a theory, we concluded that is because of differences in
spatial orientation of the CRDs. We have shown that FCN-MBL76
binds the best to a sugar, mannan.
[0074] Examples of useful fusion proteins are presented in FIG.
3. In our test molecules, FCN-MBL126 has only the carbohydrate
recognition domain (CRD) from MBL and the rest includes the
MASP-binding site. The amino-terminus is from L-FCN.
[0075] FCN-MBL76 has the CRD, neck and part of the flanking
sequences of the MASP-binding site from MBL; the lysine and
other flanking sequences of the MASP-binding site, and the
amino-terminal is from L-FCN.
[0076] FCN-MBL64 has the CRD, neck, MASP-binding site and
flanking sequences and the "kink" from MBL; the amino-terminal
is from L-FCN.
[0077] In one embodiment, one uses a fusion protein which
includes the signal peptide from L-FCN because this component is
important to signal the protein to be transported from the
cytosol to the endoplasmic reticulum for packaging and
secretion.
[0078] Accordingly, based on the description herein and
throughout this specification and examples, a skilled artisan
can design various fusion proteins, including proteins with
stability-increasing modifications using routine methods.
[0079] In certain embodiments, the methods of the present
invention include treatment and/or prevention of infections
including bacterial, viral and fungal infections. The viral
infections according to the present invention can be caused by
any virus, such as viruses including but not limited to the
viruses of the herpes family, such as Herpes Simpex I, Herpes
Simplex II, Human Herpesvirus 6 (HHV-6), herpes zoster;
poxviruses; corona viruses; paramyxoviruses; and togaviruses,
HIV, Ebola, and the like.
[0080] In certain embodiments, the methods of the present
invention provide for treatment of bacterial infections and/or
preventing bacterial infection for bacteria such as
Staphylococcus spp., Streptococcus spp., Escherichia spp.,
Enterococcus spp., Pseudomonas spp. bacteria and combinations
thereof, and more particularly Staphylococcus aureus, including
antibiotic resistant strains such as methicillin resistant
Staphylococcus aureus, Staphylococcus epidermidis, Escherichia
coli (E. coli), Pseudomonas aeruginosa (Pseudomonasae),
Streptococcus pyogenes, and combinations thereof.
[0081] In certain embodiments, the method of the present
invention provide treatment and/or prevention for infections
caused by Staphylococcus aureus; Neisseria meningitidis;
Burkholderia multivorans, group B streptococcus, Escherichia
coli, Pseudomonas aeruginosa, Mycoplasma pneumoniae, and
Chlamydia pneumoniae.
[0082] In one embodiment, the method of the present invention
provide treatment and/or prevention for infections caused by
HIV, influenza, severe acute respiratory syndrome coronavirus
SARS-CoV), hepatitis B virus, hepatitis C virus, respiratory
syncytial virus, herpes simplex virus, or filovirus, for example
Ebola or Marburg virus.
[0083] A medicament comprising MBL and/or MBL-ficolin fusion
protein, may be produced by using the eluant obtained from the
affinity chromatography as such. It is however preferred that
the eluant is subjected to further purification steps before
being used in a pharmaceutically acceptable carrier.
[0084] In one embodiment, the composition or medicament consists
essentially of MBL and/or MBL-ficolin fusion protein or
functional, i.e. infectious agents binding derivatives thereof
in a pharmaceutically acceptable carrier.
[0085] In addition to the MBL oligomers or ficolin-MBL fusion
proteins, the medicament may comprise a pharmaceutically
acceptable carrier substance and/or vehicles. In particular, a
stabilizing agent may be added to stabilize the MBL proteins or
the ficolin-MBL fusion proteins. The stabilizing agent may be a
sugar alcohol, saccharides, proteins and/or amino acids.
Examples of stabilizing agents are maltose or albumin.
[0086] The term "derivative" as used herein refers to MBL or
ficolin-MBL fusion proteins which are functional in the sense
that they can bind infectious agents but have also have been
chemically modified, for example but not limited to by
techniques such as ubiquitination, labeling, pegylation
(derivatization with polyethylene glycol, PEG) or addition of
other molecules. A molecule also a "derivative" of another
molecule when it contains additional chemical moieties not
normally a part of the molecule. Such moieties can improve the
molecule's solubility, absorption, biological half life, etc.
The moieties can alternatively decrease the toxicity of the
molecule, eliminate or attenuate any undesirable side effect of
the molecule, etc. Moieties capable of mediating such effects
are disclosed in Remington's Pharmaceutical Sciences, 18th
edition, A. R. Gennaro, Ed., MackPubl., Easton, Pa. (1990), and
PROTEINS-STRUCTURE AND MOLECULAR PROPERTIES, 2nd Ed., T. E.
Creighton, W.H. Freeman and Company, New York (1993);
POSTTRANSLATIONAL COVALENT MODIFICATION OF PROTEINS, B. C.
Johnson, Ed., Academic Press, New York, pgs. 1-12 (1983);
Seifter et al., Meth Enzymol 182:626-646 (1990); Rattan et al.,
Ann NY Acad Sci 663:48-62 (1992).).
[0087] In one embodiment, the MBL and/or ficolin-MBL fusion
protein is fused to a second fusion partner, such as a carrier
molecule to enhance its bioavailability. Such carriers are known
in the art and include poly (alkyl) glycol such as poly ethylene
glycol (PEG). Fusion to serum albumin can also increase the
serum half-life of therapeutic polypeptides.
[0088] The MBL and/or ficolin MBL fusion polypeptide can also be
fused to a second fusion partner, for example, to a polypeptide
that targets the product to a desired location, or, for example,
a tag that facilitates its purification, if so desired. Tags and
fusion partners can be designed to be cleavable, if so desired.
Another modification specifically contemplated is attachment,
e.g., covalent attachment, to a polymer. In one aspect, polymers
such as polyethylene glycol (PEG) or methoxypolyethylene glycol
(mPEG) can increase the in vivo half-life of proteins to which
they are conjugated. Methods of PEGylation of polypeptide agents
are well known to those skilled in the art, as are
considerations of, for example, how large a PEG polymer to use.
[0089] As used herein, the term "conjugate" or "conjugation"
refers to the attachment of two or more entities to form one
entity. For example, the methods of the present invention
provide conjugation of a MBL or ficolin-MBL fusion polypeptide
or fragments, derivatives or variants thereof, joined with
another entity, for example a moiety such as a first fusion
partner that makes the MBL or ficolin-MBL fusion protein stable,
such as Ig carrier particle, for example IgG1 Fc. The attachment
can be by means of linkers, chemical modification, peptide
linkers, chemical linkers, covalent or non-covalent bonds, or
protein fusion or by any means known to one skilled in the art.
The joining can be permanent or reversible. In some embodiments,
several linkers can be included in order to take advantage of
desired properties of each linker and each protein in the
conjugate. Flexible linkers and linkers that increase the
solubility of the conjugates are contemplated for use alone or
with other linkers as disclosed herein. Peptide linkers can be
linked by expressing DNA encoding the linker to one or more
proteins in the conjugate. Linkers can be acid cleavable,
photocleavable and heat sensitive linkers. Methods for
conjugation are well known by persons skilled in the art and are
encompassed for use in the present invention.
[0090] According to the present invention, the MBL or
ficolin-MBL fusion polypeptide or fragments, derivatives or
variants thereof, can be linked to the first fusion partner via
any suitable means, as known in the art, see for example U.S.
Pat. Nos. 4,625,014, 5,057,301 and 5, 514,363, which are
incorporated herein in their entirety by reference. For example,
the MBL or ficolin-MBL fusion polypeptide can be covalently
conjugated to the IgG1 Fc, either directly or through one or
more linkers. In one embodiment, a MBL or ficolin-MBL fusion
polypeptide as disclosed herein is conjugated directly to the
first fusion partner (e.g. Fc), and in an alternative
embodiment, a MBL or ficolin-MBL fusion polypeptide as disclosed
herein can be conjugated to a first fusion partner (such as IgG1
Fc) via a linker, e.g. a transport enhancing linker.
[0091] As used herein, the term "treating" includes reducing or
alleviating at least one adverse effect or symptom of an
infection. Accordingly, the anti-viral medicament according to
the present invention may be a medicament capable of virus
attenuation and/or elimination. Similarly, antibacterial
medicament according to the present invention may be a
medicament capable of stabilizing the bacterial infection and/or
eliminating such an infection.
[0092] The MBL or ficolin-MBL fusion protein can be administered
by any appropriate route which results in an effective treatment
of an infection in the subject. In one embodiment, the
administration is performed systemically.
[0093] In one embodiment, one administers the MBL or ficolin-MBL
fusion proteins enterally, topically or parenterally. The
phrases "parenteral administration" and "administered
parenterally" as used herein means modes of administration other
than enteral and topical administration, usually by injection,
and includes, without limitation, intravenous, intramuscular,
intraarterial, intrathecal, intraventricular, intracapsular,
intraorbital, intracardiac, intradermal, intraperitoneal,
transtracheal, subcutaneous, subcuticular, intraarticular, sub
capsular, subarachnoid, intraspinal, intracerebro spinal, and
infrasternal injection and infusion. The phrases "systemic
administration," "administered systemically", "peripheral
administration" and "administered peripherally" as used herein
mean the administration of MBL and/or ficolin-MBL fusion protein
other than directly into the central nervous system, such that
it enters the animal's system and, thus, is subject to
metabolism and other like processes, for example, subcutaneous
administration.
[0094] The phrase "pharmaceutically acceptable" is employed
herein to refer to those compounds, materials, compositions,
and/or dosage forms which are, within the scope of sound medical
judgment, suitable for use in contact with the tissues of human
beings and animals without excessive toxicity, irritation,
allergic response, or other problem or complication,
commensurate with a reasonable benefit/risk ratio.
[0095] The phrase "pharmaceutically acceptable carrier" as used
herein means a pharmaceutically acceptable material, composition
or vehicle, such as a liquid or solid filler, diluent,
excipient, solvent or encapsulating material, involved in
carrying or transporting the subject agents from one organ, or
portion of the body, to another organ, or portion of the body.
Each carrier must be "acceptable" in the sense of being
compatible with the other ingredients of the formulation.
[0096] Other conventional additives may be added to the
medicament depending on administration form for example. In one
embodiment the medicament is in a form suitable for injections.
Conventional carrier substances, such as isotonic saline, may be
used.
[0097] In another embodiment the pharmaceutical composition or
medicament is in a form suitable for pulmonal administration,
such as in the form of a powder for inhalation or cream or fluid
for topical application.
[0098] The route of administration may be any suitable route,
such as intravenously, intramusculary, intraperitoneally,
subcutanously or intradermally. Also, pulmonal or topical
administration is envisaged by the present invention.
[0099] The MBL composition may also be administered
simultaneously, sequentially or separately with another
anti-bacterial, anti-viral or viral or bacterial infection
symptom alleviating treatment.
[0100] The MBL and/or ficolin-MBL composition is administered in
suitable dosage regimes, in particular, it is administered
repeatedly at suitable intervals, such as once or twice a week.
For example, one can start before exposure to the virus and
maintain the periodic administration at intervals, for example
1, 2, 3, 4, 5, 6, or 7 times a week, or, for example, 1, 2, 3,
or 4 times a day, at least during a part of the exposure or
suspected exposure of the individual to the virus. One can also
begin administering the MBL composition at the time of suspected
exposure and continue with periodic additional dosages for at
least 2, 3, 4, 5, 6, or 7 days or even longer. One can also
begin the treatment at the onset of the symptoms of the
infection, such a viral infection and continue the periodic
administration of at least one additional dosages until the
symptoms begin to diminish or until there are no symptoms, or
until at least 1, 2, 3, 4, 5, 6, 7 days after the symptoms have
disappeared.
[0101] In one embodiment, the invention provides a method
wherein recombinant human MBL is administered every 1, 2, 3, 4,
5, 6, 7, 8, 9, 10 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,
22, 23, or 24 hours. In one embodiment, one administers
recombinant human MBL every 12 hours.
[0102] The use of an MBL or ficolin-MBL fusion protein
composition may also be in a kit-of-parts further comprising
another medicament, such as an anti-fungal, anti-yeast,
anti-bacterial and/or anti-viral medicament.
[0103] Accordingly, in one embodiment, the invention provides a
method for treatment and/or prevention of an infection in an
individual comprising administering to said individual a
supraphysiological amount of mannose-binding lectin (MBL) or
ficolin-MBL fusion protein or a combination thereof and a
pharmaceutically acceptable carrier. In one embodiment, the
infection is a viral infection. In one embodiment, the viral
infection is an Ebola virus infection. In another embodiment,
the infection is a bacterial infection.
[0104] In one embodiment the supraphysiological amount is an
amount that results in blood concentrations of the MBL or the
ficolin-MBL fusion protein at about 2-10 times the average
physiological MBL serum concentration. In one embodiment, the
average physiological MBL serum concentration is about 2
[mu]g/mL.
[0105] In one aspect of the invention and all other aspects
described herein, the invention provides use of
supraphysiological amount of mannose-binding lectin (MBL) or
ficolin-MBL fusion protein for treatment and/or prevention of an
infection. In one embodiment, the infection is a viral
infection. In one embodiment, the viral infection is an Ebola
virus infection. In another embodiment, the infection is a
bacterial infection.
[0106] In one aspect of the invention and all other aspects
described herein, the invention provides, one uses MBL2, SEQ ID
NO: 2, SEQ ID NO: 5, SEQ ID NO: 6 and/or SEQ ID NO: 7, or
combinations thereof in the methods of the invention. A skilled
artisan can create alternative protein variants and fusion
proteins for the use in the methods of the present invention
using routine gene manipulation techniques and sequences
available in public and proprietary databases alike.
**EXAMPLES**
[0107] We have developed novel immunotherapeutic agents that
effectively prevent and treat infections, such as
life-threatening infections, like infections caused by Ebola
(EBOV) or Marburg viruses. In our experiments we used Ebola
viruses as examples. However, based on our discovery, any other
viral or bacterial infection can be treated using similar
methods.
[0108] Ebola viruses are filamentous, enveloped, non-segmented,
negative-strand RNA viruses. EBOV subspecies Zaire and Sudan are
highly pathogenic in humans and cause as high as 90% mortality
in outbreaks in equatorial Africa. There are no FDA-approved
vaccines or therapeutic agents available to prevent or treat
EBOV.
[0109] MBL is a broad-spectrum ligand-specific C-type lectin
that plays an important role in innate immunity by acting as an
opsonin and by activating the lectin complement pathway.
Preliminary data indicated that recombinant human MBL (rhMBL) 1)
binds high-mannose residues in EBOV envelope glycoproteins (GP)
which are the principal virulence and immunogenic determinants
of EBOV, and Holmskov, U., et al., Immunol. Today 15:67-74,
1994) inhibits experimental EBOV infection in a
complement-dependent manner manner (Ji X et al. J Gen Virol
2005; 86:2535-42). These observations strongly support rhMBL's
role as a novel immunotherapeutic agent for EBOV.
[0110] We also tested three chimeric proteins (FCN-MBL)
comprising varying lengths of the carboxy terminal of MBL and
the collagen stalk of L-ficolin, another lectin-like protein
that activates the lectin complement pathway (see FIGS. 3 and
3).
[0111] RhMBL and FCN-MBL chimeras have comparable ligand-binding
specificity but FCN-MBL has a simpler multimeric structure and
different functional characteristics, and rhMBL reduces
mortality in a mouse model of native EBOV Zaire infection.
[0112] Clinical grade rhMBL was provided by ENZON
Pharmaceuticals, NJ.
[0113] Chimeric FCN-MBL proteins were expressed in stably
transfected HEK293F cells cultured in an artificial capillary
cell culture system (CELLMAX, Spectrum Laboratories, CA).
Plasmids were provided by ENZON Pharmaceuticals, NJ. Proteins
were batch purified with mannose-agarose beads, eluted with
EDTA-containing buffer, and then dialyzed with the same buffer
as for rhMBL.
[0114] The designs of the three chimeric FCN-MBL fusion proteins
are as follows:
[0115] L-FCN-MBL126: L-FCN (signal sequence+collagen+"hinge" to
ficolin domain amino acid [aa]128)+MBL (from aa126 carbohydrate
binding domain [CRD]) (SEQ ID NO: 5).
[0116] L-FCN-MBL76: L-FCN (signal sequence+part of collagen to
aa82)+MBL (from aa76 including rest of
collagen+coil-coil+carbohydrate binding domain) (SEQ ID NO: 6).
[0117] L-FCN-MBL64: L-FCN (signal sequence+part of collagen to
aa69)+MBL (from aa64 rest of collagen [containing
"kink"]+coil-coil+carbohydrate binding domain) (SEQ ID NO: 7).
[0118] Endotoxin assay: endotoxin was <5 EU/mL (FDA standard)
in all protein preparations as determined by the kinetic Limulus
amebocyte lysate test.
[0119] Ebola viruses: For in vitro experiments, HIV particles
(env-negative pNL 4-3) that lacked gp120/gp41 and that expressed
luciferase were pseudo-typed with Ebola glycoprotein. Viral
concentrations were determined by ELISA for p24 core protein.
Native EBOV subspecies Zaire was used for mouse experiments.
[0120] Structure and function of rhMBL and chimeric proteins:
the relative oligomerization was demonstrated with SDS-PAGE;
protein composition was determined by amino acid analysis.
Relative capacity to activate complement was determined by C4
deposition in an ELISA format with mannan as the capture
antigen; and relative avidity was studied with a range of
acetylated and non-acetylated carbohydrates in a competitive
ELISA format.
[0121] HEK293F infection-inhibition assay: 400 pg p24 HIV-Ebola
GP particles were preincubated with MBL-deficient serum (1:2
dilution) that was supplemented with varying amounts of rhMBL to
test the relative capacity of rhMBL to inhibit infection.
HEK293F cells (5\*10<3>/well in a 96-well format) were then
infected with the viral particles by means of spinoculation\*2
hrs and then incubated with virus-free fresh media\*40 hrs. Cell
infection was determined by expression of luciferase.
[0122] Murine model of EBOV infection: relevant PK parameters
(t1/2, Cmax) of rhMBL were calculated in 8-week old C57B/6J mice
(n=5 per group) after I.P. injection of 75 mcg (3 mg/kg) or 350
mcg (14 mg/kg). C57B/6J mice were challenged with 100pfu EBOV
Zaire (3000\*LD50) I.P. immediately after or 12 hours before
treatment with rhMBL 350 mcg I.P. that was continued every 12
hours\*10 days.
**Results**
[0123] Three chimeric proteins (FCN-MBL) were designed (FIG. 3).
The chimeric FCN-MBL proteins comprise varying lengths of the
carboxyl terminal of MBL (FIGS. 1 and 2) and the collagen stalk
of L-ficolin, another lectin-like protein that activates the
lectin complement pathway (FIG. 2). The carboxyl terminal of MBL
has been shown to be the region responsible for binding
carbohydrates, the carbohydrate recognition domain (CRD) (FIG.
2). Human MBL nucleotide reference sequence, MBL2 CDS was
derived from a consensus CDS 7247.1/NCBI NM-000242.2, the
sequence of the 747 by nucleic acid is as follows: atgtccctgt
ttccatcact ccctctcctt ctcctgagta tggtggcagc gtcttactca
gaaactgtga cctgtgagga tgcccaaaag acctgccctg cagtgattgc
ctgtagctct ccaggcatca acggcttccc aggcaaagat gggcgtgatg
gcaccaaggg agaaaagggg gaaccaggcc aagggctcag aggcttacag
ggcccccctg gaaagttggg gcctccagga aatccagggc cttctgggtc
accaggacca aagggccaaa aaggagaccc tggaaaaagt ccggatggtg
atagtagcct ggctgcctca gaaagaaaag ctctgcaaac agaaatggca
cgtatcaaaa agtggctcac cttctctctg ggcaaacaag ttgggaacaa
gttcttcctg accaatggtg aaataatgac ctttgaaaaa gtgaaggcct
tgtgtgtcaa gttccaggcc tctgtggcca cccccaggaa tgctgcagag
aatggagcca ttcagaatct catcaaggag gaagccttcc tgggcatcac
tgatgagaag acagaagggc agtttgtgga tctgacagga aatagactga
cctacacaaa ctggaacgag ggtgaaccca acaatgctgg ttctgatgaa
gattgtgtat tgctactgaa aaatggccag tggaatgacg tcccctgctc
cacctcccat ctggccgtct gtgagttccc tatctga (SEQ ID NO: 1).
[0124] Human MBL protein reference sequence, translation of MBL2
results in a 248 amino acid sequence as follows: mslfpslpll
llsmvaasys etvtcedaqk tcpaviacss pgingfpgkd grdg/kgekg
epgqglrglq gppgklgppg npgpsgspgp kgqkgdpgks pdgdsslaas
erkalqtema rikkwltfsl gkqvgnkffl tngeimtfek vkalcvkfqa
svatprnaae ngaiqnlike eaflgitdek tegqfvdltg nrltytnwne
gepnnagsde dCvlllkngq wndvpcstsh lavcefpi (SEQ ID NO: 2).
[0125] Human L-ficolin nucleotide reference sequence FCN2 CDS
was derived from consensus 6983.1/NCBI NM-004108.2. The sequence
of this 942 nucleic acid sequence is as follows:
[0000]
(SEQ ID NO: 3)
atggagctgg acagagctgt gggggtcctg ggcgctgcca
ccctgctgct ctctttcctg ggcatggcct gggctctcca
ggcggcagac acctgtccag aggtgaagat ggtgggcctg
gagggctctg acaagctcac cattctccga ggctgtccgg
ggctgcctgg ggcccctggg cccaagggag aggcaggca
caatggaaag agaggagaac gtggcccccc tggacctcct
gggaaggcag gaccacctgg gcccaacgga gcacctgggg
agccccagcc gtgcctgaca ggcccgcgta cctgcaagga
cctgctagac cgagggcact tcctgagcgg ctggcacacc
atctacctgc ccgactgccg gcccctgact gtgctctgtg
acatggacac ggacggaggg ggctggaccg ttttccagcg
gagggtggat ggctctgtgg acttctaccg ggactgggcc
acgtacaagc agggcttcgg cagtcggctg ggggagttct
ggctggggaa tgacaacatc cacgccctga ccgcccaggg
aaccagcgag ctccgtgtag acctggtgga ctttgaggac
aactaccagt ttgctaagta cagatcattc aaggtggccg
acgaggcgga gaagtacaat ctggtcctgg gggccttcgt
ggagggcagt gcgggagatt ccctgacgtt ccacaacaac
cagtccttct ccaccaaaga ccaggacaat gatcttaaca
ccggaaattg tgctgtgatg tttcagggag cttggtggta
caaaaactgc catgtgtcaa acctgaatgg tcgctacctc
agggggactc atggcagctt tgcaaatggc atcaactgga
agtcggggaa aggatacaat tatagctaca aggtgtcaga
gatgaaggtg cgacctgcct ag.
[0126] Human L-ficolin protein reference sequence translation of
FCN2, isoform a, results in a 313 amino acid protein: meldravgvl
gaatlllsfl gmawalqaad tcpevkmvgl egsdkltilr gcpglpgapg
pkgeagtngk rgergppgpp gkagppgpng apgepqpclt gprtckdlld
rghflsgwht iylpdcrplt vlcdmdtdgg gwtvfqrrvd gsvdfyrdwa
tykqgfgsrl gefwlgndni haltaqgtse lrvdlvdfed nyqfakyrsf
kvadeaekyn lylgafvegs agdsltfhnn qsfstkdqdn dlntgncavm
fqgawwyknc hvsnlngryl rgthgsfang inwksgkgyn ysykvsemkv rpa (SEQ
ID NO: 4).
[0127] Human H-ficolin nucleotide reference sequence, FCN3 CDS
transcript variant 1 derived from consensus CDS 300.1/NCBI
NM-003665.2, comprises a 990 by sequence as follows:
[0000]
(SEQ ID NO: 8)
atggatctac tgtggatcct gccctccctg tggcttctcc
tgcttggggg gcctgcctgcctgaagaccc aggaacaccc
cagctgccca ggacccaggg aactggaagc
cagcaaagttgtcctcctgc ccagttgtcc cggagctcca
ggaagtcctg gggagaaggg agccccaggtcctcaagggc
cacctggacc accaggcaag atgggcccca agggtgagcc
aggagatccagtgaacctgc tccggtgcca ggaaggccc
agaaactgcc gggagctgtt gagccagggcgccaccttga
gcggctggta ccatctgtgc ctacctgagg gcagggccct
cccagtcttttgtgacatgg acaccgaggg gggcggctgg
ctggtgtttc agaggcgcca ggatggttctgtggatttct
tccgctcttg gtcctcctac agagcaggtt ttgggaacca
agagtctgaattctggctgg gaaatgagaa tttgcaccag
cttactctcc agggtaactg ggagctgcgggtagagctgg
aagactttaa tggtaaccgt actttcgccc actatgcgac
cttccgcctcctcggtgagg tagaccacta ccagctggca
ctgggcaagt tctcagaggg cactgcaggggattccctga
gcctccacag tgggaggccc tttaccacct atgacgctga
ccacgattcaagcaacagca actgtgcagt gattgtccac
ggtgcctggt ggtatgcatc ctgttaccga tcaaatctca
atggtcgcta tgcagtgtct gaggctgccg cccacaaata
tggcattgactgggcctcag gccgtggtgt gggccacccc
taccgcaggg ttcggatgat gcttcgatag.
[0128] Human H-ficolin protein reference sequence of 299 amino
acids (translation of FCN3 transcript variant 1) is as follows:
mdllwilpsl wllllggpac lktqehpscp gpreleaskv vllpscpgap
gspgekgapgpqgppgppgk mgpkgepgdp vnllrcqegp rncrellsqg atlsgwyhlc
1pegralpvf cdmdtegggw lvfqrrqdgs vdffrswssy ragfgnqese
fwlgnenlhq ltlqgnwelrveledfngnr tfahyatfrl lgevdhyqla lgkfsegtag
dslslhsgrp fttydadhds snsncavivh gawwyascyr snlngryays
eaaahkygid wasgrgvghp yrrvrmmlr (SEQ ID NO: 9).
[0129] The sequence of the L-ficolin MBL fusion proteins used in
the experiments is set forth as follows:
[0130] L-FCN-MBL126, a 251 amino acid protein:
[0000]
(SEQ ID NO: 5)
MELDRAVGVLGAATLLLSFLGMAWALQAADTCPEVKMVGLEGSDKLTILR
GCPGLPGAPGPKGEAGTNGKRGERGPPGPPGKAGPPGPNGAPGEPQPCLT
GPRTCKDLLDRGHFLSGWHTIYLPDCRPLTFSLGKQVGNKFFLTNGEIMT
FEKVKALCVKFQASVATPRNAAENGAIQNLIKEEAFLGITDEKTEGQFVD
LTGNRLTYTNWNEGEPNNAGSDEDCVLLLKNGQWNDVPCSTSHLAVCEFP
I.
[0131] L-FCN-MBL76, a 255 amino acid protein:
[0000]
(SEQ ID NO: 6)
MELDRAVGVLGAATLLLSFLGMAWALQAADTCPEVKMVGLEGSDKLTILR
GCPGLPGAPGPKGEAGTNGKRGERGPPGPPGKLGPPGNPGPSGSPGPKGQ
KGDPGKSPDGDSSLAASERKALQTEMARIKKWLTFSLGKQVGNKFFLTNG
EIMTFEKVKALCVKFQASVATPRNAAENGAIQNLIKEEAFLGITDEKTEG
QFVDLTGNRLTYTNWNEGEPNNAGSDEDCVLLLKNGQWNDVPCSTSHLAV
CEFPI.
[0132] L-FCN-MBL64, a 254 amino acid protein:
[0000]
(SEQ ID NO: 7)
MELDRAVGVLGAATLLLSFLGMAWALQAADTCPEVKMVGLEGSDKLTILR
GCPGLPGAPGPKGEAGTNGQGLRGLQGPPGKLGPPGNPGPSGSPGPKGQK
GDPGKSPDGDSSLAASERKALQTEMARIKKWLTFSLGKQVGNKFFLTNGE
IMTFEKVKALCVKFQASVATPRNAAENGAIQNLIKEEAFLGITDEKTEGQ
FVDLTGNRLTYTNWNEGEPNNAGSDEDCVLLLKNGQWNDVPCSTSHLAVC
EFPI.
[0133] In the above-identified sequences, the protein part
indicated in bold indicates L-ficolin signal sequence; italics
indicates L-ficolin component of fusion protein; and the
remaining part is MBL component of fusion protein. The
double-underlined K indicates a Lysine=MBL-associated serine
protease (MASP) binding site.
[0134] The fusion protein numbers used herein refer to the
corresponding amino acid from the MBL protein sequence (1-248).
Accordingly, in the L-FCN-MBL126, the number 126 corresponds to
the first amino acid (L) of the MBL component of this fusion
protein (aa126-248 is the Carbohydrate Recognition Domain)
L-ficolin component=aa1-128; in the L-FCN-MBL76, the number 76
corresponds to the first amino acid (L) of the MBL component of
this fusion protein (aa126-248 is the Carbohydrate Recognition
Domain) L-ficolin component=aa1-82; and in the L-FCN-MBL64, the
number 64 corresponds to the first amino acid (Q) of the MBL
component of this fusion protein (aa126-248 is the Carbohydrate
Recognition Domain) L-ficolin component=aa1-64.
[0135] The chimeric FCN-MBL fusion proteins were expressed,
purified and analyzed on a 4-20% gradient SDS-PAGE gel, which
was stained with Imperial Blue after electrophoresis. The
recombinant chimeric proteins were compared to the full length
rhMBL. An aliquot of 450 ng of each of the recombinant proteins
was separated under reducing conditions. On the gel, the
chimeric proteins exhibited an apparent molecular weight of ~30
kDa, slightly smaller than the full length rhMBL (FIG. 4). Only
a single polypeptide was expressed for each construct.
[0136] Under non-reducing conditions (1200 ng purified protein),
the recombinant chimeric proteins primarily form trimers and
tetramers whereas rhMBL forms larger multimers (FIG. 5). The
full-length rhMBL forms larger multimers than 3 chimeric
proteins that comprise varying lengths of the carboxy-terminal
of MBL and the amino-terminal of L-ficolin.
[0137] Binding of the rhMBL or any of the three chimeric FCN-MBL
fusion proteins (10 mcg/mL in a 96-well ELISA format) to mannan
was competed with mannan and then detected with anti-hMBL mAb
(131-01). Binding to mannan was similar for all proteins except
in a narrow range of concentrations (FIG. 6). The chimeric
proteins bind a similar spectrum of carbohydrate ligands as
demonstrated in the competitive ELISA assay.
[0138] FIG. 13 shows a Kaplan Meier survival curve with a
post-challenge analysis which demonstrated that recombinant
human MBL-treated wild-type mice had a significant survival
advantage: 40% survived compared to 100% mortality among
wild-type and C3 knock-out mice treated with saline or rhMBL
indicating that rhMBL provides protection but that the
protection is dependent on C3. MBL treated mice survived
significantly longer than mice not treated with MBL. EBOV was
administered N 100pfu (plaque forming units) 3000xLD50. WT
(wildtype, C57B/6J mice) versus C3 knock out (KO). Recombinant
MBL (rhMBL) was administered at 350 mcg IL 12 hors post
challenge, then q12 h\*10 days vs. sham Rx. \*log rank,
p<0.0004.
[0139] The rhMBL and the three chimeric FCN-MBL fusion proteins
exhibited similar functional capacity to activate complement as
determined by C4 deposition. FIG. 7 shows the C4 deposition
assay that compared the capacity of rhMBL and the chimeric
proteins to bind C4. This test indicates the complement
activating activity of lectins. We showed that FCN-MBL76 has
significantly greater C4 binding activity compared with rhMBL
and the other chimerics.
[0140] Calreticulin binding assay. The 96-well ELISA plate was
coated with rhMBL or chimeric proteins (10 ug/mL), blocked with
BSA and incubated with 5 ug/mL biotinylated human placental
calreticulin that was measured at absorbance O.D. 405. FCN-MBL76
bound to human placental calreticulin significantly better than
rhMBL or the other chimeric proteins. This likely has important
implications for the relative functions of the proteins because
calreticulin is the putative cellular receptor on phagocytes for
native MBL and therefore, enhanced binding of the chimeric
molecule results in improved pathogen clearance by
opsonophagocytosis (FIG. 8).
[0141] Inhibition assay using Hep G2 cells infected with
lentivirus (HIV) pseudotyped with Ebola glycoprotein. Hep G2
cells at approximately 80% confluence in 96-well tissue culture
plates were infected with HIV particles without an envelope
(HIV-env neg; solid square) or with an envelope consisting of
Ebola glycoprotein (other symbols). The virions encoded
luciferase that was expressed only in infected cells and
detected with a commercial luciferase assay. Before addition of
viral particles to the cells, the viruses were preincubated with
0, 0.1 or 1 ug/mL of rhMBL or chimeric proteins in
veronal-buffered saline with 5 mM CaCl2 for 1 hour at 37 C.
Infection was achieved by spinoculation of cells at 1000 g\*2
hrs. The viral protein mixture was replaced with EMEM culture
media and incubated at 37 C for 40 hrs after which, the cells
were lyzed and luciferase expression was quantified. rhMBL and
the chimeric proteins inhibited viral infection to similar
significant extents (1 ug/mL vs no protein, p<0.001) (FIG.
9).
[0142] The pharmacokinetic modeling of rhMBL in immunocompetent
C57B/6J mice revealed that doses of 75 mcg and 350 mcg doses
produced Cmax of ~5 mcg/mL and ~15 mcg/mL, respectively and
half-life of ~11 hours at both doses (FIG. 10). A previous study
showed that 75 mcg is the minimum dose of rhMBL required to
activate complement in an MBL-deficient mouse model.
[0143] Using the higher dosage of 350 mcg/ml, the survival rate
of mice infected with the EBOV Zaire virus was analyzed. Data is
presented as the Kaplan Meier survival analyses. 350 mcg rhMBL
was given immediately pre-challenge with EBOV Zaire and
continued every 12 hrs\*10 days. These group of mice had a 42%
survival rate (log rank, p<0.008). When 350 mcg rhMBL was
given 12 hours post-challenge with EBOV Zaire and continued
every 12 hrs\*10 days, the mice also had a 42% survival rate (log
rank, p<0.002). Therefore, the MBL proteins have
preventive/prophylactic function as well as treatment function
against viral infections.
[0144] Accordingly, our data clearly demonstrate that rhMBL and
chimeric FCN-MBL proteins activate complement (bind C4) to a
similar extent. Since rhMBL has a half-life in mice of -11
hours, supraphysiologic doses of rhMBL administered in
prophylactic and therapeutic regimens every 12 hours
significantly reduced mortality by >40%.
[0145] The references cited herein and throughout the
specification and examples are herein incorporated by reference
in their entirety.
---
**US8394764**
**GRIFFITHSIN, GLYCOSYLATION-RESISTANT
GRIFFITHSIN, AND RELATED CONJUGATES, COMPOSITIONS, NUCLEIC
ACIDS, VECTORS, HOST CELLS, METHODS OF PRODUCTION AND
METHODS OF USE**
An isolated and purified nucleic acid molecule that encodes a
polypeptide comprising at least eight contiguous amino acids of
SEQ ID NO: 3, wherein the at least eight contiguous amino acids
have anti-viral activity, as well as an isolated and purified
nucleic acid molecule that encodes a polypeptide comprising at
least eight contiguous amino acids of SEQ ID NO: 3, wherein the
at least eight contiguous amino acids have anti-viral activity,
and, when the at least eight contiguous amino acids comprise
amino acids 1-121 of SEQ ID NO:; 3, the at least eight
contiguous amino acids have been rendered
glycosylation-resistant, a vector comprising such an isolated
and purified nucleic acid molecule, a host cell comprising the
nucleic acid molecule, optionally in the form of a vector, a
method of producing an anti-viral polypeptide or conjugate
thereof, the anti-viral polypeptide itself, a conjugate or
fusion protein comprising the anti-viral polypeptide, and
compositions comprising an effective amount of the anti-viral
polypeptide or conjugate or fusion protein thereof. Further
provided are methods of inhibiting prophylactically or
therapeutically a viral infection of a host.
**TECHNICAL FIELD OF THE INVENTION**
[0004] The invention relates to an anti-viral polypeptide, a
glycosylation-resistant anti-viral polypeptide, and related
conjugates, compositions, nucleic acids, vectors, host cells,
antibodies and methods of production and use.
**BACKGROUND OF THE INVENTION**
[0005] The field of viral therapeutics has developed in response
to the need for agents effective against retroviruses,
especially HIV. There are many ways in which an agent can
exhibit anti-retroviral activity (e.g., see DeClercq, Adv. Virus
Res., 42: 1-55 (1993); DeClercq, J. Acquir. Immun. Def. Synd.,
4: 207-218 (1991); and Mitsuya et al., Science, 249: 1533-1544
(1990). Nucleoside derivatives, such as AZT, which inhibit the
viral reverse transcriptase, were among the first clinically
active agents available commercially for anti-HIV therapy.
Although very useful in some patients, the utility of AZT and
related compounds is limited by toxicity and insufficient
therapeutic indices for fully adequate therapy. Also, given the
subsequent revelations about the true dynamics of HIV infection
(Coffin, Science, 267: 483-489 (1995); and Cohen, Science, 267:
179 (1995)), it has become increasingly apparent that agents
acting as early as possible in the viral replicative cycle are
needed to inhibit infection of newly produced, uninfected immune
cells generated in the body in response to the virus-induced
killing of infected cells. Also, it is essential to neutralize
or inhibit new infectious virus produced by infected cells.
[0006] Effective means for preventing HIV infection also are
needed as a global priority. Heterosexual transmission accounts
for the majority of new cases of HIV infection each year.
Current reports from the World Health Organization estimate that
a total of more than 40 million people are now infected with
HIV. HIV prevention research has to date focused predominantly
on vaccine development. However, no effective preventative or
therapeutic vaccine has been identified thus far. New approaches
to vaccine development, as well as entirely different strategies
and agents for preventing person-to-person transmission of HIV
infection, are needed. One approach showing great promise is the
development and use of topical microbicides. In this approach, a
suitable antiviral agent is applied directly at the potential
site of virus exposure, e.g., the genital mucosa in the case of
HIV. A suitable antiviral agent is one which inactivates or
inhibits infectivity of a virus upon contact of the antiviral
agent with the virus. Suitable animal models are available for
demonstrating in vivo efficacy of such approaches for preventing
transmission of immunodeficiency viruses, such as HIV. For
instance, the HIV-inactivating protein, cyanovirin-N, has been
shown to inhibit the sexual transmission of a chimeric
simian/human immunodeficiency virus (SHIV) infection in a
primate model employing macaques exposed to the virus vaginally
or rectally (C-C Tsai et al., AIDS Res. Hum. Retroviruses, 19,
535-541 (2003) and C-C Tsai et al., AIDS Res. Hum. Retroviruses,
20, 11-18 (2004)).
[0007] Infection of people by influenza viruses is also a major
cause of pandemic illness, morbidity and mortality worldwide.
The adverse economic consequences, as well as human suffering,
are enormous. Available treatments for established infection by
this virus are either minimally effective or ineffective; these
treatments employ amantatadine, rimantadine and neuraminidase
inhibitors. Of these drugs, only the neuraminidase inhibitors
are substantially active against multiple strains of influenza
virus that commonly infect humans, yet these drugs still have
limited utility or efficacy against pandemic disease.
[0008] Currently, the only effective preventative treatment
against influenza viral infection is vaccination. However, this,
like the drug treatments, is severely limited by the propensity
of influenza viruses to mutate rapidly by genetic exchange,
resulting in the emergence of highly resistant viral strains
that rapidly infect and spread throughout susceptible
populations. In fact, a vaccination strategy is only effective
from year-to-year if the potential pandemic strains can be
identified or predicted, and corresponding vaccines prepared and
administered early enough that the year's potential pandemic can
be aborted or attenuated. Thus, new preventative and therapeutic
interventions and agents are urgently needed to combat influenza
viruses.
[0009] New agents with broad anti-influenza virus activity
against diverse strains, clinical isolates and subtypes of
influenza virus would be highly useful, since such agents would
most likely remain active against the mutating virus. The two
major types of influenza virus that infect humans are influenza
A and B, both of which cause severe acute illness that may
include both respiratory and gastrointestinal distress, as well
as other serious pathological sequellae. An agent that has
anti-influenza virus activity against diverse strains and
isolates of both influenza A and B, including recent clinical
isolates thereof, would be particularly advantageous for use in
prevention or treatment of hosts susceptible to influenza virus
infection.
[0010] The predominant mode of transmission of influenza viral
infection is respiratory, i.e., transmission via inhalation of
virus-laden aerosolized particles generated through coughing,
sneezing, breathing, etc., of an influenza-infected individual.
Transmission of infectious influenza virions may also occur
through contact (e.g., through inadvertent hand-to-mouth
contact, kissing, touching, etc.) with saliva or other bodily
secretions of an infected individual. Thus, the primary first
points of contact of infectious influenza virions within a
susceptible individual are the mucosal surfaces within the
oropharyngeal mucosa, and the mucosal surfaces within the upper
and lower respiratory tracts. Not only do these sites comprise
first points of virus contact for initial infection of an
individual, they are also the primary sites for production and
exit (e.g., by coughing, sneezing, salivary transmission, etc.)
of bodily fluids containing infectious influenza viral
particles. Therefore, availability of a highly potent
anti-influenza virus agent, having broad-spectrum activity
against diverse strains and isolates of influenza viruses A and
B, which could be applied or delivered topically to the
aforementioned mucosal sites of contact and infection and
transmission of infectious influenza viruses, would be highly
advantageous for therapeutic and preventative inhibition of
influenza viral infection, either in susceptible uninfected or
infected hosts.
[0011] In this regard, new classes of anti-viral agents, to be
used alone or in combination existing anti-viral agents, are
needed for effective anti-viral therapy. New agents are also
important for the prophylactic inhibition of viral infection. In
both areas of need, the ideal new agent(s) would act as early as
possible in the viral life cycle; be as virus-specific as
possible (i.e., attack a molecular target specific to the virus
but not the host); render the intact virus noninfectious;
prevent the death or dysfunction of virus-infected cells;
prevent further production of virus from infected cells; prevent
spread of virus infection to uninfected cells; be highly potent
and active against the broadest possible range of strains and
isolates of a given virus; be resistant to degradation under
physiological and rigorous environmental conditions; and be
readily and inexpensively produced.
[0012] Accordingly, the invention provides a novel anti-viral
polypeptide and related conjugates, nucleic acids, vectors, host
cells and methods of production and use. This and other
advantages of the invention, as well as additional inventive
features, will become apparent from the description provided
herein.
**BRIEF SUMMARY OF THE INVENTION**
[0013] The invention provides, among other things, an isolated
and purified nucleic acid molecule that encodes a polypeptide
comprising at least eight contiguous amino acids of SEQ ID NO:
3, wherein the at least eight contiguous amino acids have
anti-viral activity, optionally as part of an encoded fusion
protein. In this regard, the invention also provides an isolated
and purified nucleic acid molecule that encodes a polypeptide
comprising at least eight contiguous amino acids of SEQ ID NO:
3, wherein the at least eight contiguous amino acids comprise
amino acids 1-121 of SEQ ID NO: 3 which have been rendered
glycosylation-resistant and wherein the at least eight
contiguous amino acids have antiviral activity, optionally as
part of an encoded fusion protein. Further provided are vectors
comprising an aforementioned isolated and purified nucleic acid
molecule and a host cell or organism comprising such a vector.
[0014] Accordingly, the invention also provides a method of
producing an anti-viral polypeptide, which method comprises
expressing the nucleic acid molecule, optionally in the form of
a vector, in a host cell or organism. Thus, an anti-viral
polypeptide comprising at least eight contiguous amino acids of
SEQ ID NO: 3, wherein the at least eight contiguous amino acids
have anti-viral activity, and an antiviral polypeptide
comprising at least eight contiguous amino acids of SEQ ID NO:
3, wherein the at least eight contiguous amino acids comprise
amino acids 1-121 of SEQ ID NO: 3, which have been rendered
glycosylation-resistant and wherein the at least eight
contiguous amino acids have antiviral activity, are also
provided, as are conjugates comprising an aforementioned
anti-viral polypeptide and at least one effector component.
Compositions comprising an effective amount of an aforementioned
anti-viral polypeptide or anti-viral polypeptide conjugate are
also provided.
[0015] The invention further provides a method of inhibiting
prophylactically or therapeutically a viral infection of a host,
specifically a retroviral infection of a host, such as an
infection of a host with a human immunodeficiency virus (HIV),
e.g., HIV-1 or HIV-2, or influenza virus. The method comprises
administering to the host an effective amount of an anti-viral
polypeptide or anti-viral polypeptide conjugate comprising at
least eight contiguous amino acids of SEQ ID NO: 3, wherein the
at least eight contiguous amino acids have anti-viral activity,
whereupon the viral infection is inhibited.
[0016] Still further provided is a method of inhibiting a viral
infection of an animal comprising transforming host cells in
vivo with a nucleic acid molecule encoding an above-described
polypeptide. Even still further provided is a method of
inhibiting a viral infection of an animal comprising
transforming host cells with a nucleic acid molecule encoding an
above-described polypeptide and placing the transformed host
cells into or onto the animal.
[0017] An antibody that binds griffithsin is provided as is a
composition comprising same. Similarly, an anti-griffithsin
antibody is provided as is a composition comprising same. A
method of administering an anti-griffithsin antibody or a
composition comprising same to a mammal so as to inhibit
infection of the mammal with a virus is also provided.
**BRIEF DESCRIPTION OF THE DRAWINGS****[0018] FIG. 1 is a flow diagram illustrating an anti-HIV
bioassay-guided method of isolating, purifying, and
elucidating the amino acid sequence of griffithsin.****[0019] FIG. 2 is a flow diagram illustrating a method of
synthesizing a recombinant griffithsin gene.****[0020] FIG. 3 is a flow diagram illustrating a method of
expressing a synthetic griffithsin gene encoding a His-tagged
griffithsin polypeptide protein and purification of the
recombinant His-tagged griffithsin.****[0021] FIG. 4a is a line graph illustrating the anti-HIV
activity of native griffithsin, in terms of concentration of
griffithsin (nM) (X-axis) versus % control (Y-axis). FIG. 4b
is a line graph illustrating the anti-HIV activity of
recombinant, His-tagged griffithsin in terms of concentration
of griffithsin (nM) (X-axis) versus % control (Y-axis).****[0022] FIG. 5a is a bar graph comparing test proteins
bound by griffithsin (Y-axis) and absorbance of the
griffithsin-test protein complex at 405 nm (X-axis). FIG. 5b
illustrates the concentration-dependent binding of griffithsin
to glycosylated (-) or nonglycosylated () gp120 by comparing
griffithsin (GRFT) concentration (pmol) and absorbance of
griffithsin-gp120 complexes at 405 nm.****[0023] FIG. 6 is a flow diagram illustrating a method of
producing anti-griffithsin antibodies.****[0024] FIG. 7 is the amino acid sequence of griffithsin
polypeptide (SEQ ID NO: 3) isolated and purified from
Griffithsin sp.****[0025] FIG. 8 shows the nucleic acid (SEQ ID NO: 1)
sequence of recombinant griffithsin.****[0026] FIG. 9 is the amino acid sequence of a recombinant
griffithsin polypeptide (SEQ ID NO: 2).****[0027] FIG. 10 shows the nucleic acid sequence of a
recombinant griffithsin polypeptide comprising a His tag (SEQ
ID NO: 4).****[0028] FIG. 11 is the amino acid sequence of a
recombinant griffithsin polypeptide comprising a His tag (SEQ
ID NO: 5).** **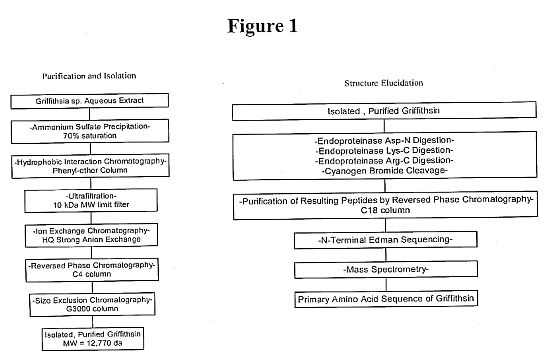  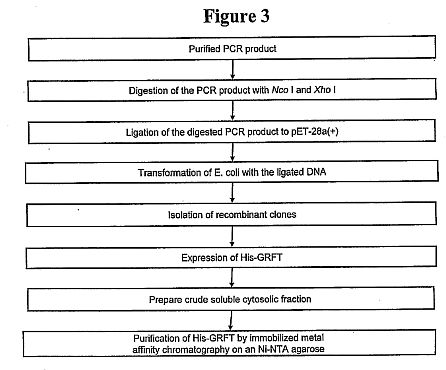 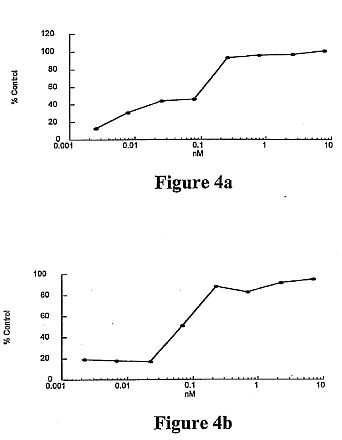 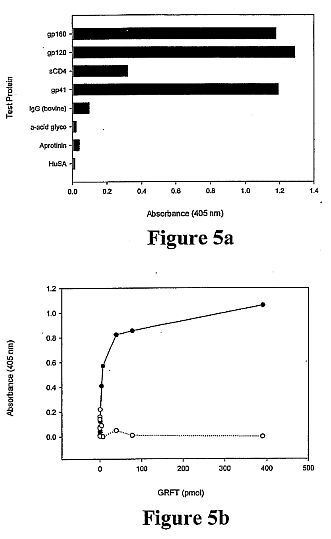      ****DETAILED DESCRIPTION OF THE PRESENT INVENTION**
[0029] The principal overall objective of the invention is to
provide an anti-viral polypeptide and derivatives thereof, and
broad uses thereof (e.g., medical and research uses), including
prophylactic and/or therapeutic applications against viruses. An
initial observation, which led to the invention, was anti-viral
activity of certain extracts from a marine organism, namely
Rhodophyte (Griffithsia sp.), originally collected in the
territorial waters of New Zealand. Low picomolar concentrations
of a protein isolated from the extracts, referred to herein as
griffithsin, irreversibly inactivated human clinical isolates of
HIV. Its HIV molecular target is high mannose-comprised
oligosaccharide constituents of Env glycoproteins. Upon binding,
griffithsin inhibits viral binding, fusion, and entry.
Griffithsin also targets other viruses having oligosaccharide
constituents similar to HIV, such as other retroviruses, e.g.,
FIV, SIV and HTLV, and non-retroviruses, e.g., influenza, ebola,
and measles.
[0030] Accordingly, the invention provides an isolated and
purified anti-viral polypeptide of SEQ ID NO: 3 from Griffithsia
sp. and functional homologs thereof, referred to collectively as
"griffithsin." Herein the term "griffithsin" is used generically
to refer to a natural griffithsin or any related, functionally
equivalent (i.e., anti-viral) polypeptide or derivative thereof.
By definition, in this context, a related, functionally
equivalent polypeptide or derivative thereof (a) contains a
sequence of at least eight contiguous amino acids directly
identical to a sub-sequence of eight contiguous amino acids
contained within a natural griffithsin, and (b) can specifically
bind to a virus, in particular an influenza virus or a
retrovirus, more specifically a primate immunodeficiency virus,
more specifically HIV-1, HIV-2 or SIV, or to an infected host
cell expressing one or more viral antigen(s), more specifically
an envelope glycoprotein, such as gp120, of the respective
virus. In addition, such a functionally equivalent polypeptide
or derivative thereof can comprise the amino acid sequence of a
natural griffithsin (see SEQ ID NO: 3), in which 1-20,
preferably 1-10, more preferably 1, 2, 3, 4, or 5, and most
preferably 1 or 2, amino acids have been removed from one or
both ends, preferably from only one end, e.g., removed from the
amino-terminal end, of natural griffithsin. Alternatively, a
functionally equivalent polypeptide or derivative thereof can
comprise the amino acid sequence of a native griffithsin (see
SEQ ID NO: 3), in which 1-20, preferably 1-10, more preferably
1, 2, 3, 4, or 5, and most preferably 1 or 2, amino acids have
been added to one or both ends, preferably from only one end,
e.g., the amino-terminal end, of the native griffithsin.
[0031] The invention further provides an isolated and purified
polypeptide encoded by a nucleic acid molecule comprising a
sequence of SEQ ID NO: 1 or a nucleic acid molecule encoding an
amino acid sequence of SEQ ID NO: 2 or SEQ ID NO: 3. Upon
examination of the antiviral griffithsin polypeptide, the amino
acid at position 31 of SEQ ID NO: 3 (represented as Xaa) was
found not to be a familiar amino acid residue. Placement of an
alanine at position 31, such as achieved in the recombinant
griffithsin polypeptide described herein (SEQ ID NO: 2), results
in a polypeptide exhibiting equivalent activity as the natural
griffithsin polypeptide. If desired, the amino acid at position
31 can be substituted with any other amino acid to facilitate
protein production. Ideally, the substitution at position 31 of
SEQ ID NO: 3 does not diminish the anti-viral activity of the
protein (e.g., does not diminish the anti-viral activity more
than 50%, more than 30% or more than 10%) as compared to the
anti-viral activity of the native protein. Preferably, the
aforementioned nucleic acid molecules encode at least eight
(e.g., at least 10, at least 20, at least 30, at least 50, at
least 70, at least 80, at least 90, or at least 100) contiguous
amino acids of the amino acid sequence of SEQ ID NO: 3, which
desirably have anti-viral activity. If the at least eight
contiguous amino acids of SEQ ID NO: 3 comprise amino acids
1-121, desirably amino acid residue 45, 60, 71, and/or 104 has
been rendered glycosylation resistant, while maintaining
antiviral activity of the polypeptide.
[0032] The term "isolated" as used herein means having been
removed from its natural environment. The term "purified" as
used herein means having been increased in purity, wherein
"purity" is a relative term and not to be construed as absolute
purity. By "antiviral" is meant that the polypeptide or fragment
thereof can inhibit a virus (e.g., inhibit entry of a virus into
a host cell, limit the spread of viral infection by inhibiting
cell to cell fusion, and the like), in particular an influenza
virus, such as influenza virus or a strain A or strain B, or a
retrovirus, specifically a primate immunodeficiency virus, more
specifically a human immunodeficiency virus (HIV), such as
HIV-1, HIV-2 or SIV.
[0033] Preferably, the polypeptide or derivative thereof
comprises an amino acid sequence that is substantially
homologous to that of an anti-viral protein from Griffithsia sp.
By "substantially homologous" is meant sufficient homology to
render the polypeptide or derivative thereof anti-viral, with
anti-viral activity characteristic of an anti-viral protein
isolated from Griffithsia sp. At least about 50% homology (e.g.,
at least about 60% homology, at least about 65% homology, or at
least about 70% homology), preferably at least about 75%
homology (e.g., at least about 80% homology or at least about
85% homology), and most preferably at least about 90% homology
(e.g., at least about 95% homology) should exist.
[0034] Alterations of the natural amino acid sequence to produce
variant polypeptides can be done by a variety of means known to
those skilled in the art. For instance, amino acid substitutions
can be conveniently introduced into the polypeptides at the time
of synthesis. Alternatively, site-specific mutations can be
introduced by ligating into an expression vector a synthesized
oligonucleotide comprising the modified site. Alternately,
oligonucleotide-directed, site-specific mutagenesis procedures
can be used, such as disclosed in Walder et al., Gene, 42: 133
(1986); Bauer et al., Gene, 37: 73 (1985); Craik, Biotechniques,
12-19 (January 1995); and U.S. Pat. Nos. 4,518,584 and
4,737,462.
[0035] It is within the skill of the ordinary artisan to select
synthetic and naturally-occurring amino acids that effect
conservative or neutral substitutions for any particular
naturally-occurring amino acids. The ordinarily skilled artisan
desirably will consider the context in which any particular
amino acid substitution is made, in addition to considering the
hydrophobicity or polarity of the side-chain, the general size
of the side chain and the pK value of side-chains with acidic or
basic character under physiological conditions. For example,
lysine, arginine, and histidine are often suitably substituted
for each other, and more often arginine and histidine. As is
known in the art, this is because all three amino acids have
basic side chains, whereas the pK value for the side-chains of
lysine and arginine are much closer to each other (about 10 and
12) than to histidine (about 6). Similarly, glycine, alanine,
valine, leucine, and isoleucine are often suitably substituted
for each other, with the proviso that glycine is frequently not
suitably substituted for the other members of the group. This is
because each of these amino acids are relatively hydrophobic
when incorporated into a polypeptide, but glycine's lack of an
[alpha]-carbon allows the phi and psi angles of rotation (around
the [alpha]-carbon) so much conformational freedom that glycinyl
residues can trigger changes in conformation or secondary
structure that do not often occur when the other amino acids are
substituted for each other. Other groups of amino acids
frequently suitably substituted for each other include, but are
not limited to, the group consisting of glutamic and aspartic
acids; the group consisting of phenylalanine, tyrosine and
tryptophan; and the group consisting of serine, threonine and,
optionally, tyrosine. Additionally, the ordinarily skilled
artisan can readily group synthetic amino acids with
naturally-occurring amino acids.
[0036] The ordinarily skilled artisan can generate griffithsin
mutants or variants by, for example, substituting or mutating
amino acids which are not critical for the anti-viral function
of the polypeptide. Ideally, mutations that do not modify the
electronic or structural environment of the peptide are
generated to retain optimal antiviral activity. For example,
natural griffithsin forms dimers, which can be advantageous in
some embodiments. Therefore, alterations which do not disrupt
dimer formation can be preferred. Amino acid residues which are
not responsible for folding or stability of the
three-dimensional conformation of the griffithsin polypeptide
are candidate residues for mutation. Alternatively or in
addition, amino acids which are not involved in glycoprotein
binding can be mutated or replaced. It is understood that
surface hydrophobicity plays a key role in protein-protein
interactions and surface electrophilicity is important to
protein-sugar interactions, such as the interaction between
griffithsin and viral proteins. Hydrophobic surface clusters and
electrophilic surface clusters on the griffithsin peptide or
homologs which suggest regions critical for interaction with the
viral envelope can be mapped using routine methods such as those
disclosed in Bewley et al., Nature Structural Biology, 5(7):
571-578 (1998). Amino acid residues not found either in
electrophilic or hydrophobic surface clusters are likely not
critical for hydrophobicity or electrophilicity of these
clusters and, thus, are appropriate targets for mutation to
create griffithsin fragments (e.g., anti-viral polypeptides
comprising at least about eight contiguous amino acids of SEQ ID
NO: 2 or SEQ ID NO: 3), variants, mutants, or homologs (e.g.,
griffithsin variants having 80%, 85%, or 90% homology to SEQ ID
NO: 2 or SEQ ID NO: 3) which retain antiviral activity. If
desired, amino acid residues which are responsible for binding
to high-mannose oligosaccharide-containing glycoproteins on the
viral surface can be mutated to increase the specificity or
affinity of glycoprotein binding.
[0037] If desired, the proteins and peptides of the invention
(including antiviral fragments, variant polypeptides, fusion
proteins, and conjugates) can be modified, for instance, by
glycosylation, amidation, carboxylation, or phosphorylation, or
by the creation of acid addition salts, amides, esters, in
particular C-terminal esters, and N-acyl derivatives of the
proteins of the invention. The polypeptides also can be modified
to create protein derivatives by forming covalent or noncovalent
complexes with other moieties in accordance with methods known
in the art. Covalently-bound complexes can be prepared by
linking the chemical moieties to functional groups on the side
chains of amino acids comprising the proteins, or at the N- or
C-terminus. Desirably, such modifications and conjugations do
not adversely affect the activity of the polypeptides (and
variants thereof). While such modifications and conjugations can
have greater or lesser activity, the activity desirably is not
negated and is characteristic of the unaltered polypeptide.
[0038] The polypeptides (and fragments, homologs, variants, and
fusion proteins) can be prepared by any of a number of
conventional techniques. The polypeptide can be isolated or
purified from a naturally occurring source or from a recombinant
source. For instance, in the case of recombinant proteins, a DNA
fragment encoding a desired polypeptide can be subcloned into an
appropriate vector using well-known molecular genetic techniques
(see, e.g., Maniatis et al., Molecular Cloning: A Laboratory
Manual, 2nd ed. (Cold Spring Harbor Laboratory (1989)) and other
references cited herein under "EXAMPLES"). The fragment can be
transcribed and the polypeptide subsequently translated in
vitro. Commercially available kits also can be employed (e.g.,
such as manufactured by Clontech, Palo Alto, Calif.; Amersham
Life Sciences, Inc., Arlington Heights, Ill.; InVitrogen, San
Diego, Calif.; and the like). The polymerase chain reaction
optionally can be employed in the manipulation of nucleic acids.
[0039] Such polypeptides also can be synthesized using an
automated peptide synthesizer in accordance with methods known
in the art. Alternately, the polypeptide (and fragments,
homologs, variants, and fusion proteins) can be synthesized
using standard peptide synthesizing techniques well-known to
those of skill in the art (e.g., as summarized in Bodanszky,
Principles of Peptide Synthesis, (Springer-Verlag, Heidelberg:
1984)). In particular, the polypeptide can be synthesized using
the procedure of solid-phase synthesis (see, e.g., Merrifield,
J. Am. Chem. Soc., 85: 2149-54 (1963); Barany et al., Int. J.
Peptide Protein Res., 30: 705-739 (1987); and U.S. Pat. No.
5,424,398). If desired, this can be done using an automated
peptide synthesizer. Removal of the t-butyloxycarbonyl (t-BOC)
or 9-fluorenylmethyloxycarbonyl (Fmoc) amino acid blocking
groups and separation of the polypeptide from the resin can be
accomplished by, for example, acid treatment at reduced
temperature. The protein-containing mixture then can be
extracted, for instance, with diethyl ether, to remove
non-peptidic organic compounds, and the synthesized polypeptide
can be extracted from the resin powder (e.g., with about 25% w/v
acetic acid). Following the synthesis of the polypeptide,
further purification (e.g., using HPLC) optionally can be
preformed in order to eliminate any incomplete proteins,
polypeptides, peptides or free amino acids. Amino acid and/or
HPLC analysis can be performed on the synthesized polypeptide to
validate its identity. For other applications according to the
invention, it may be preferable to produce the polypeptide as
part of a larger fusion protein, either by chemical conjugation
or through genetic means, such as are known to those skilled in
the art. In this regard, the invention also provides a fusion
protein comprising the isolated or purified antiviral
polypeptide (or fragment thereof) or variant thereof and one or
more other protein(s) having any desired properties or effector
functions, such as cytotoxic or immunological properties, or
other desired properties, such as to facilitate isolation,
purification, analysis, or stability of the fusion protein.
[0040] A griffithsin conjugate comprising a griffithsin coupled
to at least one effector component, which can be the same or
different, is also provided. The effector component can be
polyethylene glycol, dextran, albumin, an immunological reagent,
a toxin, an antiviral agent, or a solid support matrix.
"Immunological reagent" will be used to refer to an antibody, an
antibody fragment (e.g., an F(ab')2, an Fab', an Fab, an Fv, an
sFv, a dsFv, or an Fc antibody fragment), an immunoglobulin, and
an immunological recognition element. An immunological
recognition element is an element, such as a peptide, e.g., the
FLAG sequence of a recombinant griffithsin-FLAG fusion protein,
which facilitates, through immunological recognition, isolation
and/or purification and/or analysis of the protein or peptide to
which it is attached. An immunological reagent also can be an
immunogenic peptide, which can be fused to griffithsin for
enhancing an immune response. In this respect, the invention
provides an anti-viral conjugate comprising a griffithsin
polypeptide or fragment thereof bound to a virus or viral
envelope glycoprotein. A griffithsin fusion protein is a type of
griffithsin conjugate, wherein a griffithsin is coupled to one
or more other protein(s) having any desired properties or
effector functions, such as cytotoxic or immunological
properties, or other desired properties, such as to facilitate
isolation, purification or analysis of the fusion protein or
increase the stability or in vivo half-life of the fusion
protein. Griffithsin also can be attached to a chemical moiety
which allows recognition, isolation, purification, and/or
analysis of the protein or peptide. An example of such a
chemical moiety is a His tag of a recombinant griffithsin-His
fusion protein.
[0041] A "toxin" can be, for example, Pseudomonas exotoxin. An
"antiviral agent" can be AZT, ddI, ddC, 3TC gancyclovir,
fluorinated dideoxynucleosides, nevirapine, R82913, Ro 31-8959,
BI-RJ-70, acyclovir, [alpha]-interferon, recombinant sCD4,
michellamines, calanolides, nonoxynol-9, gossypol and
derivatives thereof, gramicidin, amantatadine, rimantadine, and
neuraminidase inhibitors, and cyanovirin-N or a functional
homolog or derivative thereof (see, for example, U.S. Pat. No.
5,843,882). A "solid support matrix" can be a magnetic bead, a
flow-through matrix, a sponge, a stent, a culture plate, or a
matrix comprising a contraceptive device, such as a condom,
diaphragm, cervical cap, vaginal ring or contraceptive sponge.
In an alternative embodiment, a solid support matrix can be an
implant for surgical implantation in a host and, if appropriate,
later removal.
[0042] In view of the foregoing, the invention further provides
a composition comprising (i) the isolated or purified antiviral
polypeptide (or fragment thereof), a variant thereof, a fusion
protein of the antiviral polypeptide (or fragment thereof) or
variant thereof, and a conjugate of the antiviral polypeptide
(or fragment thereof) or variant thereof, and/or (ii) a carrier,
excipient or adjuvant therefor. Preferably, component (i) of the
composition is present in an antiviral effective amount and the
carrier is pharmaceutically acceptable. By "antiviral effective
amount" is meant an amount sufficient to inhibit the infectivity
of the virus.
[0043] The carrier can be any of those conventionally used and
is limited only by chemico-physical considerations, such as
solubility and lack of reactivity with the active agent of the
invention, and by the route of administration. It is preferred
that the pharmaceutically acceptable carrier be one which is
chemically inert to the active agent and one which has no
detrimental side effects or toxicity under the conditions of
use. The pharmaceutically acceptable carriers described herein,
for example, vehicles, adjuvants, excipients, and diluents, are
well-known to those ordinarily skilled in the art and are
readily available to the public. Typically, the composition,
such as a pharmaceutical composition, can comprise a
physiological saline solution; dextrose or other saccharide
solution; or ethylene, propylene, polyethylene, or other glycol.
The pharmaceutical composition preferably does not comprise
mannose or N-acetyl-glucosamine, as these molecules may
interfere with the functioning of the antiviral agent.
[0044] The invention also provides a method of obtaining a
griffithsin from Griffithsia sp. Such a method comprises (a)
identifying an extract of Griffithsia sp. containing anti-viral
activity, (b) optionally removing high molecular weight
biopolymers from the extract, (c) anti-viral bioassay-guided
fractionating the extract to obtain a crude extract of
griffithsin, and (d) purifying the crude extract by
reverse-phase HPLC to obtain griffithsin (see, also, Example 1).
More specifically, the method involves the use of ethanol to
remove high molecular weight biopolymers from the extract and
the use of an anti-HIV bioassay to guide fractionation of the
extract.
[0045] Griffithsin (a polypeptide of exactly SEQ ID NO: 3),
which was isolated and purified using the aforementioned method,
was subjected to conventional procedures typically used to
determine the amino acid sequence of a given pure protein. Thus,
the griffithsin was initially sequenced by N-terminal Edman
degradation of intact protein and numerous overlapping peptide
fragments generated by endoproteinase digestion. Amino acid
analysis was in agreement with the deduced sequence. ESI mass
spectrometry of reduced, HPLC-purified griffithsin showed a
molecular ion consistent with the calculated value. These
studies indicated that griffithsin from Griffithsia was
comprised of a unique sequence of 121 amino acids having little
or no significant homology or identity to previously described
proteins or transcription products of known nucleotide
sequences. No more than eight contiguous amino acids from
griffithsin were found in any amino acid sequences from known
proteins, nor were there any known proteins from any source
having significant sequence identity with griffithsin. Given the
chemically deduced amino acid sequence of griffithsin, a
corresponding recombinant griffithsin (r-griffithsin) was
created and used to establish definitively that the deduced
amino acid sequence was, indeed, active against virus, such as
HIV and influenza.
[0046] Accordingly, the invention provides isolated and purified
nucleic acid molecules and synthetic nucleic acid molecules,
which comprise a coding sequence for a griffithsin, such as an
isolated and purified nucleic acid molecule comprising a
sequence of SEQ ID NO: 1, an isolated and purified nucleic acid
molecule encoding an amino acid sequence of SEQ ID NO: 2, an
isolated and purified nucleic acid sequence encoding an amino
acid sequence SEQ ID NO: 3, an isolated and purified nucleic
acid molecule comprising a sequence of SEQ ID NO: 4, an isolated
and purified nucleic acid sequence encoding an amino acid
sequence of SEQ ID NO: 5, and a nucleic acid molecule that is
substantially homologous or substantially identical to any one
of the aforementioned nucleic acid molecules. By "substantially
homologous" is meant sufficient homology to render the
polypeptide or derivative thereof anti-viral, with anti-viral
activity characteristic of an anti-viral protein isolated from
Griffithsia. At least about 50% homology or identity (e.g., at
least about 60%, at least about 65%, or at least about 70%
homology or identity), preferably at least about 75% homology or
identity (e.g., at least about 80% or at least about 85%
homology or identity), and most preferably at least about 90%
homology or identity (e.g., at least about 95% homology or
identity) should exist.
[0047] The inventive nucleic acid molecule preferably comprises
a nucleic acid sequence encoding at least eight (preferably at
least 10, more preferably at least 20, and most preferably at
least 30) contiguous amino acids of the amino acid sequence of
SEQ ID NO: 3 or SEQ ID NO: 2. The inventive nucleic acid
molecule also comprises a nucleic acid sequence encoding a
polypeptide comprising the amino acid sequence of a native
griffithsin, in which 1-20, preferably 1-10, more preferably 1,
2, 3, 4, or 5, and most preferably 1 or 2, amino acids have been
removed from one or both ends, preferably from only one end,
e.g., removed from the amino-terminal end, of the native
griffithsin. Alternatively, the nucleic acid molecule can
comprise a nucleic acid sequence encoding a polypeptide
comprising the amino acid sequence of a natural griffithsin (see
SEQ ID NO: 3), in which 1-20, preferably 1-10, more preferably
1, 2, 3, 4, or 5, and most preferably 1 or 2, amino acids have
been added to one or both ends, preferably from only one end,
e.g., the amino-terminal end, of the native griffithsin.
Preferably, the isolated and purified nucleic acid molecule
encodes a polypeptide comprising at least eight contiguous amino
acids of SEQ ID NO: 3, which desirably have anti-viral activity.
If the at least eight contiguous amino acids comprise amino
acids 1-121 of SEQ ID NO: 3, desirably amino acids 46, 60, 71,
and/or 104 have been rendered glycosylation resistant, while
maintaining antiviral activity of the polypeptide. Deletions and
substitutions of SEQ ID NO: 2 or SEQ ID NO: 3 are within the
skill in the art.
[0048] Given the present disclosure, it will be apparent to one
skilled in the art that a partial griffithsin gene sequence will
likely suffice to code for a fully functional, i.e., anti-viral,
such as anti-influenza or anti-HIV, griffithsin. A minimum
essential DNA coding sequence(s) for a functional griffithsin
can readily be determined by one skilled in the art, for
example, by synthesis and evaluation of sub-sequences comprising
the native griffithsin, and by site-directed mutagenesis studies
of the griffithsin DNA coding sequence.
[0049] Using an appropriate DNA coding sequence, a recombinant
griffithsin can be made by genetic engineering techniques (for
general background see, e.g., Nicholl, in An Introduction to
Genetic Engineering, Cambridge University Press: Cambridge
(1994), pp. 1-5 & 127-130; Steinberg et al., in Recombinant
DNA Technology Concepts and Biomedical Applications, Prentice
Hall: Englewood Cliffs, N.J. (1993), pp. 81-124 & 150-162;
Sofer in Introduction to Genetic Engineering,
Butterworth-Heinemann, Stoneham, Mass. (1991), pp. 1-21 &
103-126; Old et al., in Principles of Gene Manipulation,
Blackwell Scientific Publishers: London (1992), pp. 1-13 &
108-221; and Emtage, in Delivery Systems for Peptide Drugs,
Davis et al., eds., Plenum Press: New York (1986), pp. 23-33).
For example, a Griffithsia gene or cDNA encoding a griffithsin
can be identified and subcloned. The gene or cDNA then can be
incorporated into an appropriate expression vector and delivered
into an appropriate polypeptide-synthesizing organism (e.g., E.
coli, S. cerevisiae, P. pastoris, or other bacterial, yeast,
insect, plant or mammalian cells), where the gene, under the
control of an endogenous or exogenous promoter, can be
appropriately transcribed and translated. Alternatively, the
expression vector can be administered to a plant or animal, for
example, for large-scale production (see, e.g., Fischer et al.,
Transgenic Res., 9 (4-5): 279-299 (2000); Fischer et al., J.
Biol. Regul. Homeost. Agents, 14: 83-92 (2000); deWilde et al.,
Plant Molec. Biol., 43: 347-359 (2000); Houdebine, Transgenic
Research, 9: 305-320 (2000); Brink et al., Theriogenology, 53:
139-148 (2000); Pollock et al., J. Immunol. Methods, 231:
147-157 (1999); Conrad et al., Plant Molec. Biol., 38: 101-109
(1998); Staub et al., Nature Biotech., 18: 333-338 (2000);
McCormick et al., PNAS USA, 96: 703-708 (1999); Zeitlin et al.,
Nature Biotech., 16: 1361-1364 (1998); Tacker et al., Microbes
and Infection, 1: 777-783 (1999); Tacket et al., Nature Med.,
4(5): 607-609 (1998); and Methods in Biotechnology, Recombinant
Proteins from Plants, Production and Isolation of Clinically
Useful Compounds, Cunningham and Porter, eds., Humana Press:
Totowa, N.J. (1998)). Such expression vectors (including, but
not limited to, phage, cosmid, viral, and plasmid vectors) are
known to those skilled in the art, as are reagents and
techniques appropriate for gene transfer (e.g., transfection,
electroporation, transduction, micro-injection, transformation,
etc.). If a griffithsin is to be recombinantly produced in
isolated eukaryotic cells or in a eukaryotic organism, such as a
plant (see above references and also Methods in Biotechnology,
Recombinant Proteins from Plants, Production and Isolation of
Clinically Useful Compounds, Cunningham and Porter, eds., Humana
Press: Totowa, N.J. (1998)), desirably the N-linked
glycosylation sites at positions 45, 60, 71, and/or 104 is
rendered glycosylation-resistant, such as in accordance with the
methods described herein. Subsequently, the recombinantly
produced polypeptide can be isolated and purified using standard
techniques known in the art (e.g., chromatography,
centrifugation, differential solubility, isoelectric focusing,
etc.), and assayed for anti-viral activity.
[0050] Alternatively, a natural griffithsin can be obtained from
Griffithsia by non-recombinant methods, and sequenced by
conventional techniques. The sequence can then be used to
synthesize the corresponding DNA, which can be subcloned into an
appropriate expression vector and delivered into a
polypeptide-producing cell for en mass recombinant production of
the desired polypeptide.
[0051] In this regard, the invention also provides a vector
comprising a DNA sequence, e.g., a Griffithsia gene sequence for
griffithsin, a cDNA encoding a griffithsin, or a synthetic DNA
sequence encoding griffithsin. The vector can be targeted to a
cell-surface receptor if so desired. A nucleic acid molecule as
described above can be cloned into any suitable vector and can
be used to transform or transfect any suitable host. The
selection of vectors and methods to construct them are commonly
known to persons of ordinary skill in the art and are described
in general technical references (see, in general, "Recombinant
DNA Part D," Methods in Enzymology, Vol. 153, Wu and Grossman,
eds., Academic Press (1987) and the references cited herein
under "EXAMPLES"). Desirably, the vector comprises regulatory
sequences, such as transcription and translation initiation and
termination codons, which are specific to the type of host
(e.g., bacterium, fungus, plant or animal) into which the vector
is to be introduced, as appropriate and taking into
consideration whether the vector is DNA or RNA. Preferably, the
vector comprises regulatory sequences that are specific to the
genus of the host. Most preferably, the vector comprises
regulatory sequences that are specific to the species of the
host.
[0052] Constructs of vectors, which are circular or linear, can
be prepared to contain an entire nucleic acid as described above
or a portion thereof ligated to a replication system functional
in a prokaryotic or eukaryotic host cell. Replication systems
can be derived from Co1E1, 2 m[mu] plasmid, [lambda], SV40,
bovine papilloma virus, and the like.
[0053] In addition to the replication system and the inserted
nucleic acid, the construct can include one or more marker
genes, which allow for selection of transformed or transfected
hosts. Marker genes include biocide resistance, e.g., resistance
to antibiotics, heavy metals, etc., complementation in an
auxotrophic host to provide prototrophy, and the like.
[0054] One of ordinary skill in the art will appreciate that any
of a number of vectors known in the art are suitable for use in
the invention. Suitable vectors include those designed for
propagation and expansion or for expression or both. Examples of
suitable vectors include, for instance, plasmids,
plasmid-liposome complexes, and viral vectors, e.g.,
parvoviral-based vectors (i.e., adeno-associated virus
(AAV)-based vectors), retroviral vectors, herpes simplex virus
(HSV)-based vectors, and adenovirus-based vectors. Any of these
expression constructs can be prepared using standard recombinant
DNA techniques described in, e.g., Sambrook et al., Molecular
Cloning: A Laboratory Manual, 2edition, Cold Spring
Harbor Laboratory Press, Cold Spring Harbor, N.Y. (1989);
Ausubel et al., Current Protocols in Molecular Biology, Greene
Publishing Associates and John Wiley & Sons, New York, N.Y.
(1994); Fischer et al., Transgenic Res., 9 (4-5): 279-299
(2000); Fischer et al., J. Biol. Regul. Homeost. Agents, 14:
83-92 (2000); deWilde et al., Plant Molec. Biol., 43: 347-359
(2000); Houdebine, Transgenic Research, 9: 305-320 (2000); Brink
et al., Theriogenology, 53: 139-148 (2000); Pollock et al., J.
Immunol. Methods, 231: 147-157 (1999); Conrad et al., Plant
Molec. Biol., 38: 101-109 (1998); Staub et al., Nature Biotech.,
18: 333-338 (2000); McCormick et al., PNAS USA, 96: 703-708
(1999); Zeitlin et al., Nature Biotech., 16: 1361-1364 (1998);
Tacker et al., Microbes and Infection, 1: 777-783 (1999); and
Tacket et al., Nature Med., 4(5): 607-609 (1998). Examples of
cloning vectors include the pUC series, the pBluescript series
(Stratagene, LaJolla, Calif.), the pET series (Novagen, Madison,
Wis.), the pGEX series (Pharmacia Biotech, Uppsala, Sweden), and
the pEX series (Clonetech, Palo Alto, Calif.). Bacteriophage
vectors, such as [lambda]GT10, [lambda]GT11, [lambda]ZapII
(Stratagene), [lambda] EMBL4, and [lambda] NM1149, also can be
used. Examples of plant expression vectors include pBI101,
pBI101.2, pBI101.3, pBI121 and pBIN19 (Clonetech, Palo Alto,
Calif.). Examples of animal expression vectors include pEUK-C1,
pMAM and pMAMneo (Clonetech).
[0055] An expression vector can comprise a native or normative
promoter operably linked to an isolated or purified nucleic acid
as described above. The selection of promoters, e.g., strong,
weak, inducible, tissue-specific and developmental-specific, is
within the skill in the art. Similarly, the combining of a
nucleic acid molecule as described above with a promoter is also
within the skill in the art.
[0056] The DNA, whether isolated and purified or synthetic, or
cDNA encoding a griffithsin can encode for either the entire
griffithsin or a portion thereof. Where the DNA or cDNA does not
comprise the entire coding sequence of the native griffithsin,
the DNA or cDNA can be subcloned as part of a gene fusion. In a
transcriptional gene fusion, the DNA or cDNA will contain its
own control sequence directing appropriate production of protein
(e.g., ribosome binding site, translation initiation codon,
etc.), and the transcriptional control sequences (e.g., promoter
elements and/or enhancers) will be provided by the vector. In a
translational gene fusion, transcriptional control sequences as
well as at least some of the translational control sequences
(i.e., the translational initiation codon) will be provided by
the vector. In the case of a translational gene fusion, a
chimeric protein will be produced.
[0057] Genes also can be constructed for specific fusion
proteins containing a functional griffithsin component plus a
fusion component conferring additional desired attribute(s) to
the composite protein. For example, a fusion sequence for a
toxin or immunological reagent can be added to facilitate
purification and analysis of the functional protein.
[0058] Genes can be specifically constructed to code for fusion
proteins, which contain a griffithsin coupled to an effector
protein, such as a toxin or immunological reagent, for specific
targeting to a virus or viral-infected cells, e.g., HIV and/or
HIV-infected cells or influenza and/or influenza-infected cells.
In these instances, the griffithsin moiety serves not only as a
neutralizing agent but also as a targeting agent to direct the
effector activities of these molecules selectively against a
given virus, such as HIV or influenza. Thus, for example, a
therapeutic agent can be obtained by combining the HIV-targeting
function or influenza-targeting function of a functional
griffithsin with a toxin aimed at neutralizing infectious virus
and/or by destroying cells producing infectious virus, such as
HIV or influenza. Similarly, a therapeutic agent can be
obtained, which combines the viral-targeting function of a
griffithsin with the multivalency and effector functions of
various immunoglobulin subclasses. Example 6 further illustrates
the viral-targeting, specifically gp120-targeting, properties of
a griffithsin.
[0059] Similar rationales underlie extensive developmental
therapeutic efforts exploiting the HIV gp120-targeting
properties of sCD4. For example, sCD4-toxin conjugates have been
prepared in which sCD4 is coupled to a Pseudomonas exotoxin
component (Chaudhary et al., in The Human Retrovirus, Gallo et
al., eds., Academic Press: San Diego, Calif. (1991), pp.
379-387; and Chaudhary et al., Nature, 335: 369-372 (1988)), or
to a diphtheria toxin component (Aullo et al., EMBO J., 11:
575-583 (1992)) or to a ricin A-chain component (Till et al.,
Science, 242: 1166-1167 (1988)). Likewise, sCD4-immunoglobulin
conjugates have been prepared in attempts to decrease the rate
of in vivo clearance of functional sCD4 activity, to enhance
placental transfer, and to effect a targeted recruitment of
immunological mechanisms of pathogen elimination, such as
phagocytic engulfment and killing by antibody-dependent
cell-mediated cytotoxicity, to kill and/or remove HIV-infected
cells and virus (Capon et al., Nature, 337: 525-531 (1989);
Traunecker et al., Nature, 339: 68-70 (1989); and Langner et al.
(1993), supra). While such CD4-immunoglobulin conjugates
(sometimes called "immunoadhesins") have, indeed, shown
advantageous pharmacokinetic and distributional attributes in
vivo, and anti-HIV effects in vitro, clinical results have been
discouraging (Schooley et al. (1990), supra; Husson et al.
(1992), supra; and Langner et al. (1993), supra). This is not
surprising since clinical isolates of HIV, as opposed to
laboratory strains, are highly resistant to binding and
neutralization by sCD4 (Orloff et al. (1995), supra; and Moore
et al. (1992), supra). The griffithsin polypeptide binds to a
wide range of sugars present on viral glycoproteins and,
therefore, can inhibit a wide range of viruses which display
those glycoproteins. The extraordinarily broad targeting
properties of a functional griffithsin to viruses, e.g., primate
retroviruses, in general, and clinical and laboratory strains,
in particular, can be especially advantageous for combining with
toxins, immunoglobulins and other selected effector proteins.
[0060] Viral-targeted conjugates can be prepared either by
genetic engineering techniques (see, for example, Chaudhary et
al. (1988), supra) or by chemical coupling of the targeting
component with an effector component. The most feasible or
appropriate technique to be used to construct a given
griffithsin conjugate or fusion protein will be selected based
upon consideration of the characteristics of the particular
effector molecule selected for coupling to a griffithsin. For
example, with a selected non-proteinaceous effector molecule,
chemical coupling, rather than genetic engineering techniques,
may be the only feasible option for creating the desired
griffithsin conjugate.
[0061] Accordingly, the invention also provides nucleic acid
molecules encoding griffithsin fusion proteins. In particular,
the invention provides a nucleic acid molecule comprising SEQ ID
NO: 4 and substantially homologous sequences thereof. Also
provided is a vector comprising a nucleic acid sequence encoding
a griffithsin fusion protein and a method of obtaining a
griffithsin fusion protein by expression of the vector encoding
a griffithsin fusion protein in a protein-synthesizing organism
as described above. Accordingly, griffithsin fusion proteins are
also provided.
[0062] In view of the above, the invention further provides an
isolated and purified nucleic acid molecule, which comprises a
griffithsin coding sequence, such as one of the aforementioned
nucleic acids, namely a nucleic acid molecule encoding an amino
acid sequence of SEQ ID NO: 2 or SEQ ID NO: 3 or a nucleic acid
molecule comprising a sequence of SEQ ID NO: 1 coupled to a
second nucleic acid encoding an effector protein. The first
nucleic acid preferably comprises a nucleic acid sequence
encoding at least eight contiguous amino acids of the amino acid
sequence of SEQ ID NO: 2 or SEQ ID NO: 3, which encodes a
functional griffithsin, and the second nucleic acid preferably
encodes an effector protein, such as a toxin or immunological
reagent as described herein.
[0063] Accordingly, the invention also further provides an
isolated and purified fusion protein encoded by a nucleic acid
molecule comprising a sequence of SEQ ID NO: 1 or a nucleic acid
molecule encoding an amino acid sequence of SEQ ID NO: 2 or SEQ
ID NO: 3, either one of which is coupled to a second nucleic
acid encoding an effector protein. Preferably, the
aforementioned nucleic acid molecules encode at least eight
contiguous amino acids of the amino acid sequence of SEQ ID NO:
2 or SEQ ID NO: 3, which desirably have anti-viral activity,
coupled to an effector molecule, such as a toxin or
immunological reagent as described above. Preferably, the
effector molecule targets a virus, more preferably HIV or
influenza, and, most preferably glycoprotein gp120 of HIV or
hemagluttinin of influenza. If the at least eight contiguous
amino acids of SEQ ID NO: 3 (or SEQ ID NO: 2) comprise amino
acids 1-121, desirably amino acids 46, 60, 71, and/or 104 have
been rendered glycosylation-resistant, yet maintain antiviral
activity by substitution of the asparagine at those positions
with, for example, an alanine or a glutamine residue.
[0064] The coupling can be effected at the DNA level or by
chemical coupling as described above. For example, a
griffithsin-effector protein conjugate of the invention can be
obtained by (a) selecting a desired effector protein or peptide;
(b) synthesizing a composite DNA coding sequence comprising a
first DNA coding sequence comprising one of the aforementioned
nucleic acid sequences, which codes for a functional
griffithsin, coupled to a second DNA coding sequence for an
effector protein or peptide, e.g., a toxin or immunological
reagent; (c) expressing said composite DNA coding sequence in an
appropriate protein-synthesizing organism; and (d) purifying the
desired fusion protein to substantially pure form.
Alternatively, a griffithsin-effector molecule conjugate of the
invention can be obtained by (a) selecting a desired effector
molecule and a griffithsin or griffithsin fusion protein; (b)
chemically coupling the griffithsin or griffithsin fusion
protein to the effector molecule; and (c) purifying the desired
griffithsin-effector molecule conjugate to substantially pure
form.
[0065] Conjugates comprising a functional griffithsin (e.g., an
anti-viral polypeptide comprising at least eight contiguous
amino acids of SEQ ID NO: 3, such as SEQ ID NO: 3, wherein the
at least eight contiguous amino acids bind to a virus, in
particular an infectious virus, such as influenza virus or HIV,
in which case the griffithsin binds to gp120 or hemagluttinin)
coupled to an anti-griffithsin antibody, a virus, a viral
glycoprotein, or at least one effector component, which can be
the same or different, such as a toxin, an immunological
reagent, an antiviral agent, or other functional reagent, can be
designed even more specifically to exploit the unique viral
targeting, e.g., gp120-targeting properties, of griffithsins.
[0066] Other functional reagents that can be used as effector
components in the inventive conjugates can include, for example,
polyethylene glycol, dextran, albumin, a solid support matrix,
and the like, whose intended effector functions may include one
or more of the following: to improve stability of the conjugate;
to increase the half-life of the conjugate; to increase
resistance of the conjugate to proteolysis; to decrease the
immunogenicity of the conjugate; to provide a means to attach or
immobilize a functional griffithsin onto a solid support matrix
(e.g., see, for example, Harris, in Poly(Ethylene Glycol)
Chemistry: Biotechnical and Biomedical Applications, Harris,
ed., Plenum Press: New York (1992), pp. 1-14). Conjugates
furthermore can comprise a functional griffithsin coupled to
more than one effector molecule, each of which, optionally, can
have different effector functions (e.g., such as a toxin
molecule (or an immunological reagent) and a polyethylene glycol
(or dextran or albumin) molecule). Diverse applications and uses
of functional proteins and peptides, such as in the present
instance a functional griffithsin, attached to or immobilized on
a solid support matrix, are exemplified more specifically for
poly(ethylene glycol) conjugated proteins or peptides in a
review by Holmberg et al. (In Poly(Ethylene Glycol) Chemistry:
Biotechnical and Biomedical Applications, Harris, ed., Plenum
Press: New York (1992), pp. 303-324). Preferred examples of
solid support matrices include magnetic beads, a flow-through
matrix, and a matrix comprising a contraceptive device, such as
a condom, a diaphragm, a cervical cap, a vaginal ring or a
sponge.
[0067] Example 4 reveals novel gp120-directed effects of
griffithsins. Solid-phase ELISA experiments show that
griffithsin is capable of global conformational effects on
gp120, as observed as a decrease of immunoreactivity at
multiple, distinct, non-overlapping epitopes.
[0068] The range of anti-viral activity of griffithsin against
diverse CD4<+>-tropic immunodeficiency virus strains in
various target cells is remarkable; virtually all tested strains
of HIV-1, HIV-2 and SIV were similarly sensitive to griffithsin;
clinical isolates and laboratory strains showed essentially
equivalent sensitivity. Cocultivation of chronically infected
and uninfected CEM-SS cells with griffithsin did not inhibit
viral replication, but did cause a concentration-dependent
inhibition of cell-to-cell fusion and virus transmission;
similar results from binding and fusion inhibition assays
employing HeLa-CD4-LTR-[beta]-galactosidase cells were
consistent with griffithsin inhibition of virus-cell and/or
cell-cell binding.
[0069] The anti-viral, e.g., anti-HIV, activity of the
griffithsins and conjugates thereof of the invention can be
further demonstrated in a series of interrelated in vitro
anti-viral assays (Gulakowski et al., J. Virol. Methods, 33:
87-100 (1991)), which accurately predict for anti-viral activity
in humans. These assays measure the ability of compounds to
prevent the replication of HIV and/or the cytopathic effects of
HIV on human target cells. These measurements directly correlate
with the pathogenesis of HIV-induced disease in vivo. The
results of the analysis of the anti-viral activity of
griffithsins or conjugates, as set forth in Examples 5-7 and 9,
predict accurately the anti-viral activity of these products in
vivo in humans and, therefore, establish the utility of the
invention. Furthermore, since the invention also provides
methods of ex vivo use of griffithsins and conjugates, the
utility of griffithsins and conjugates thereof is even more
certain.
[0070] The griffithsins and conjugates thereof of the invention
can be shown to inhibit a virus, specifically a retrovirus, more
specifically an immunodeficiency virus, such as the human
immunodeficiency virus, i.e., HIV-1 or HIV-2. The griffithsins
and conjugates of the invention can be used to inhibit other
retroviruses as well as other viruses (see, e.g., Principles of
Virology: Molecular Biology, Pathogenesis, and Control, Flint et
al., eds., ASM Press: Washington, D.C. (2000), particularly
Chapter 19). Examples of viruses that may be treated in
accordance with the invention include, but are not limited to,
Type C and Type D retroviruses, HTLV-1, HTLV-2, HIV, FIV, FLV,
SIV, MLV, BLV, BIV, equine infectious virus, anemia virus, avian
sarcoma viruses, such as Rous sarcoma virus (RSV), hepatitis
type A, B, non-A and non-B viruses, arboviruses, varicella
viruses, human herpes virus (e.g., HHV-6), measles, mumps,
filovirus (e.g., Ebola, such as Ebola strains Sudan, Zaire, Cote
d'Ivoire, and Reston) and rubella viruses. Griffithsins and
conjugate thereof also can be used to inhibit influenza viral
infection (see, e.g., Fields Virology, third edition, Fields et
al., eds., Lippincott-Raven Publishers: Philadelphia, Pa.
(1996), particularly Chapter 45) prophylactically and
therapeutically in accordance with the methods set forth herein.
[0071] Thus, the invention further provides a composition
comprising (i) one or more of an above-described purified or
isolated nucleic acid or variant thereof, optionally as part of
an encoded fusion protein, and (ii) a carrier, excipient or
adjuvant. Preferably, (i) is present in an antiviral effective
amount and the composition is pharmaceutically acceptable. The
composition can further comprise at least one additional active
agent, such as an antiviral agent other than a griffithsin (or
antiviral fragment, fusion protein or conjugate thereof), in an
antiviral effective amount. Suitable antiviral agents include
AZT, ddA, ddI, ddC, 3TC gancyclovir, fluorinated
dideoxynucleosides, acyclovir, [alpha]-interferon, nonnucleoside
analog compounds, such as nevirapine (Shih et al., PNAS, 88:
9878-9882, (1991)), TIBO derivatives, such as R82913 (White et
al., Antiviral Res., 16: 257-266 (1991)), Ro31-8959, BI-RJ-70
(Merigan, Am. J. Med., 90 (Suppl. 4A): 8S-17S (1991)),
michellamines (Boyd et al., J. Med. Chem., 37: 1740-1745 (1994))
and calanolides (Kashman et al., J. Med. Chem., 35: 2735-2743
(1992)), nonoxynol-9, gossypol and derivatives, gramicidin,
Enfurtide (i.e., T20), cyanovirin-N and functional homologs
thereof (Boyd et al. (1997), supra). Other exemplary antiviral
compounds include protease inhibitors (see R. C. Ogden and C. W.
Flexner, eds., Protease Inhibitors in AIDS Therapy, Marcel
Dekker, N Y (2001)), such as saquinavir (see I. B. Duncan and S.
Redshaw, in R. C. Ogden and C. W. Flexner, supra, pp. 27-48),
ritonavir (see D. J. Kempf, in R. C. Ogden and C. W. Flexner,
supra, pp. 49-64), indinavir (see B. D. Dorsey and J. P. Vacca,
in R. C. Ogden and C. W. Flexner, supra, pp. 65-84), nelfinavir
(see S. H. Reich, in R. C. Ogden and C. W. Flexner, supra, pp.
85-100), amprenavir (see R. D. Tung, in R. C. Ogden and C. W.
Flexner, supra, pp. 101-118), and anti-TAT agents. If the
composition is to be used to induce an immune response, it
comprises an immune response-inducing amount of the inventive
agent and can further comprise an immunoadjuvant, such as
polyphosphazene polyelectrolyte.
[0072] The pharmaceutical composition can contain other
pharmaceuticals, such as virucides, immunomodulators,
immunostimulants, antibiotics and absorption enhancers.
Exemplary immunomodulators and immunostimulants include various
interleukins, sCD4, cytokines, antibody preparations, blood
transfusions, and cell transfusions. Exemplary antibiotics
include antifungal agents, antibacterial agents, and
anti-Pneumocystitis carnii agents. Exemplary absorption
enhancers include bile salts and other surfactants, saponins,
cyclodextrins, and phospholipids (Davis (1992), supra).
[0073] An isolated cell comprising an above-described purified
or isolated nucleic acid or variant thereof, optionally in the
form of a vector, which is optionally targeted to a cell-surface
receptor, is also provided. Examples of host cells include, but
are not limited to, a human cell, a human cell line, E. coli, B.
subtilis, P. aerugenosa, S. cerevisiae, and N. crassa. E. coli,
in particular E. coli TB-1, TG-2, DH5[alpha], XL-Blue MRF'
(Stratagene), SA2821 and Y1090. Preferably, the cell is a
mammalian cell, bacterium, or yeast. A preferred bacterium is
lactobacillus or other commensal microorganism. The
above-described nucleic acid or variant thereof, optionally in
the form of a vector, can be introduced into a host cell using
such techniques as transfection, electroporation, transduction,
micro-injection, transformation, and the like.
[0074] Accordingly, the invention provides a method of
inhibiting prophylactically or therapeutically a viral
infection, in particular an influenza viral infection or HIV
infection, of a host. The method comprises administering to the
host an effective amount of an anti-viral polypeptide or
anti-viral polypeptide conjugate comprising at least eight
contiguous amino acids of SEQ ID NO: 3, wherein the at least
eight contiguous amino acids are nonglycosylated and have
anti-viral activity, whereupon the viral infection is inhibited.
The anti-viral polypeptide can be derived from a griffithsin
obtained from Griffithsia or recombinantly produced in
accordance with the methods described above. Nonglycosylated
anti-viral polypeptides can be produced in prokaryotic
cells/organisms. Amino acids 45, 60, 71, and/or 104 in such
nonglycosylated antiviral polypeptides can be deleted or
substituted, for example, with alanine or glutamine.
Nonglycosylated antiviral polypeptides also can be produced in
eukaryotic cells/organisms by expressing a portion of a
griffithsin, such as that of SEQ ID NO: 3, that does not contain
a glycosylation site or all or a portion of a griffithsin, such
as that of SEQ ID NO: 3, which contains a glycosylation site
that has been rendered glycosylation-resistant as described and
exemplified herein. When the viral infection is an influenza
viral infection and the anti-viral polypeptide or anti-viral
polypeptide conjugate is administered topically to the host,
preferably the anti-viral protein or anti-viral peptide is
administered to the respiratory system of the host, preferably
as an aerosol or microparticulate powder.
[0075] The prophylactic and therapeutic treatment of many viral
infections, including influenza virus infections, is complicated
by appearance of virus forms resistant to currently employed
medications, such as neurominidase inhibitors. The inventive
method is particularly useful in this context, as the inventive
anti-viral polypeptide or anti-viral polypeptide conjugate binds
a wide range of glycoproteins present on the viral surface.
Accordingly, the inventive anti-viral polypeptide or conjugate
thereof can be administered to an animal, preferably a human,
dog, cat, bird, cow, pig, horse, lamb, mouse, or rat, in
combination with other anti-viral agents to guard against the
propagation of anti-viral-resistant strains of virus. In
addition, it is thought that during adaptive mutation (e.g.,
resistance to neuraminidase inhibitors), the level of
glycosylation found at the viral surface increases in some
viruses, such as influenza. Thus, in that the inventive
anti-viral agent binds sugars of viral surface glycoproteins,
the inventive method provides a valuable complimentary therapy
to current anti-viral regimens.
[0076] Griffithsins and conjugates thereof collectively comprise
polypeptides and proteins, and, as such, are particularly
susceptible to hydrolysis of amide bonds (e.g., catalyzed by
peptidases) and disruption of essential disulfide bonds or
formation of inactivating or unwanted disulfide linkages (Carone
et al., J. Lab. Clin. Med., 100:1-14 (1982)). There are various
ways to alter molecular structure, if necessary, to provide
enhanced stability to the griffithsin or conjugate thereof
(Wunsch, Biopolymers, 22: 493-505 (1983); and Samanen, in
Polymeric Materials in Medication, Gebelein et al., eds., Plenum
Press: New York (1985) pp. 227-242), which may be essential for
preparation and use of pharmaceutical compositions containing
griffithsins or conjugates thereof for therapeutic or
prophylactic applications against viruses, e.g., HIV. Possible
options for useful chemical modifications of a griffithsin or
conjugate include, but are not limited to, the following
(adapted from Samanen, J. M. (1985) supra): (a) olefin
substitution, (b) carbonyl reduction, (c) D-amino acid
substitution, (d) N-methyl substitution, (e) C-methyl
substitution, (f) C-C'-methylene insertion, (g) dehydro amino
acid insertion, (h) retro-inverso modification, (i) N-terminal
to C-terminal cyclization, and (j) thiomethylene modification.
Griffithsins and conjugates thereof also can be modified by
covalent attachment of carbohydrate and polyoxyethylene
derivatives, which are expected to enhance stability and
resistance to proteolysis (Abuchowski et al., in Enzymes as
Drugs, Holcenberg et al., eds., John Wiley: New York (1981), pp.
367-378).
[0077] Other important general considerations for design of
delivery strategy systems and compositions, and for routes of
administration, for protein and peptide drugs, such as
griffithsins and conjugates thereof (Eppstein, CRC Crit. Rev.
Therapeutic Drug Carrier Systems, 5: 99-139 (1988); Siddiqui et
al., CRC Crit. Rev. Therapeutic Drug Carrier Systems, 3: 195-208
(1987); Banga et al., Int. J. Pharmaceutics, 48: 15-50 (1988);
Sanders, Eur. J. Drug Metab. Pharmacokinetics, 15: 95-102
(1990); and Verhoef, Eur. J. Drug Metab. Pharmacokinetics, 15:
83-93 (1990)), also apply. The appropriate delivery system for a
given griffithsin or conjugate thereof will depend upon its
particular nature, the particular clinical application, and the
site of drug action. As with any protein or peptide drug, oral
delivery of a griffithsin or a conjugate thereof will likely
present special problems, due primarily to instability in the
gastrointestinal tract and poor absorption and bioavailability
of intact, bioactive drug therefrom. Therefore, especially in
the case of oral delivery, but also possibly in conjunction with
other routes of delivery, it will be necessary to use an
absorption-enhancing agent in combination with a given
griffithsin or conjugate thereof A wide variety of
absorption-enhancing agents have been investigated and/or
applied in combination with protein and peptide drugs for oral
delivery and for delivery by other routes (Verhoef (1990),
supra; van Hoogdalem, Pharmac. Ther., 44: 407-443 (1989); and
Davis, J. Pharm. Pharmacol, 44 (Suppl. 1): 186-190 (1992)). Most
commonly, typical enhancers fall into the general categories of
(a) chelators, such as EDTA, salicylates, and N-acyl derivatives
of collagen, (b) surfactants, such as lauryl sulfate and
polyoxyethylene-9-lauryl ether, (c) bile salts, such as
glycholate and taurocholate, and derivatives, such as
taurodihydrofusidate, (d) fatty acids, such as oleic acid and
capric acid, and their derivatives, such as acylcarnitines,
monoglycerides and diglycerides, (e) non-surfactants, such as
unsaturated cyclic ureas, (f) saponins, (g) cyclodextrins, and
(h) phospholipids.
[0078] Other approaches to enhancing oral delivery of protein
and peptide drugs, such as the griffithsins and conjugates
thereof, can include aforementioned chemical modifications to
enhance stability to gastrointestinal enzymes and/or increased
lipophilicity. Alternatively, or in addition, the protein or
peptide drug can be administered in combination with other drugs
or substances, which directly inhibit proteases and/or other
potential sources of enzymatic degradation of proteins and
peptides. Yet another alternative approach to prevent or delay
gastrointestinal absorption of protein or peptide drugs, such as
griffithsins or conjugates, is to incorporate them into a
delivery system that is designed to protect the protein or
peptide from contact with the proteolytic enzymes in the
intestinal lumen and to release the intact protein or peptide
only upon reaching an area favorable for its absorption. A more
specific example of this strategy is the use of biodegradable
microcapsules or microspheres, both to protect vulnerable drugs
from degradation, as well as to effect a prolonged release of
active drug (Deasy, in Microencapsulation and Related Processes,
Swarbrick, ed., Marcell Dekker, Inc.: New York (1984), pp. 1-60,
88-89, 208-211). Microcapsules also can provide a useful way to
effect a prolonged delivery of a protein and peptide drug, such
as a griffithsin or conjugate thereof, after injection
(Maulding, J. Controlled Release, 6: 167-176 (1987)).
[0079] Given the aforementioned potential complexities of
successful oral delivery of a protein or peptide drug, it is
fortunate that there are numerous other potential routes of
delivery of a protein or peptide drug, such as a griffithsin or
conjugate thereof. These routes include topical, subcutaneous,
intravenous, intraarterial, intrathecal, intracisternal, buccal,
rectal, nasal, pulmonary, transdermal, vaginal, ocular, and the
like (Eppstein (1988), supra; Siddiqui et al. (1987), supra;
Banga et al. (1988), supra; Sanders (1990), supra; Verhoef
(1990), supra; Barry, in Delivery Systems for Peptide Drugs,
Davis et al., eds., Plenum Press: New York (1986), pp. 265-275;
and Patton et al., Adv. Drug Delivery Rev, 8: 179-196 (1992)).
With any of these routes, or, indeed, with any other route of
administration or application, a protein or peptide drug, such
as a griffithsin or conjugate thereof, may initiate an
immunogenic reaction. In such situations it may be necessary to
modify the molecule in order to mask immunogenic groups. It also
can be possible to protect against undesired immune responses by
judicious choice of method of formulation and/or administration.
For example, site-specific delivery can be employed, as well as
masking of recognition sites from the immune system by use or
attachment of a so-called tolerogen, such as polyethylene
glycol, dextran, albumin, and the like (Abuchowski et al.
(1981), supra; Abuchowski et al., J. Biol. Chem., 252: 3578-3581
(1977); Lisi et al., J. Appl. Biochem, 4: 19-33 (1982); and
Wileman et al., J. Pharm. Pharmacol, 38: 264-271 (1986)). Such
modifications also can have advantageous effects on stability
and half-life both in vivo and ex vivo.
[0080] Procedures for covalent attachment of molecules, such as
polyethylene glycol, dextran, albumin and the like, to proteins,
such as griffithsins or conjugates thereof, are well-known to
those skilled in the art, and are extensively documented in the
literature (e.g., see Davis et al., in Peptide and Protein Drug
Delivery, Lee, ed., Marcel Dekker: New York (1991), pp.
831-864).
[0081] Other strategies to avoid untoward immune reactions also
can include the induction of tolerance by administration
initially of only low doses. In any event, it will be apparent
from the present disclosure to one skilled in the art that for
any particular desired medical application or use of a
griffithsin or conjugate thereof, the skilled artisan can select
from any of a wide variety of possible compositions, routes of
administration, or sites of application, what is advantageous.
[0082] Accordingly, the anti-viral griffithsins and conjugates
thereof of the invention can be formulated into various
compositions for use, for example, either in therapeutic
treatment methods for infected individuals, or in prophylactic
methods against viral, e.g., HIV and influenza virus, infection
of uninfected individuals.
[0083] The invention also provides a composition, such as a
pharmaceutical composition, which comprises an isolated and
purified griffithsin, a griffithsin conjugate, a matrix-anchored
griffithsin or a matrix-anchored griffithsin conjugate, such as
an anti-viral effective amount thereof. The composition can
further comprise a carrier, such as a pharmaceutically
acceptable carrier. The composition can further comprise at
least one additional anti-viral compound other than a
griffithsin or conjugate thereof, such as in an anti-viral
effective amount of an anti-viral compound. Suitable anti-viral
compounds include cyanovirin, AZT, ddI, ddC, gancyclovir,
fluorinated dideoxynucleosides, nevirapine, R82913, Ro 31-8959,
BI-RJ-70, acyclovir, [alpha]-interferon, recombinant sCD4,
michellamines, calanolides, nonoxynol-9, gossypol and
derivatives thereof, neuroamidase inhibitors, amantatadine,
rimantadine, enfurtide, and gramicidin. If the composition is to
be used to induce an immune response, it comprises an immune
response-inducing amount of a griffithsin or conjugate thereof
and can further comprise an immunoadjuvant, such as
polyphosphazene polyelectrolyte. The griffithsin used in the
composition, e.g., pharmaceutical composition, can be isolated
and purified from nature or genetically engineered. Similarly,
the griffithsin conjugate can be genetically engineered or
chemically coupled.
[0084] The inventive compositions can be administered to a host,
such as a human, so as to inhibit viral infection in a
prophylactic or therapeutic method. The compositions of the
invention are particularly useful in inhibiting the growth or
replication of a virus, such as influenza virus or a retrovirus,
in particular an influenza virus or an immunodeficiency virus,
such as HIV, specifically HIV-1 and HIV-2, inhibiting
infectivity of the virus, inhibiting the binding of virus to a
host cell, and the like. The compositions are useful in the
therapeutic or prophylactic treatment of animals, such as
humans, who are infected with a virus or who are at risk for
viral infection, respectively. The compositions also can be used
to treat objects or materials, such as medical equipment,
supplies, or fluids, including biological fluids, such as blood,
blood products and vaccine formulations, cells, tissues and
organs, to remove or inactivate virus in an effort to prevent or
treat viral infection of an animal, such as a human. Such
compositions also are useful to prevent sexual transmission of
viral infections, e.g., HIV, which is the primary way in which
the world's AIDS cases are contracted (Merson (1993), supra).
Adherence of the inventive anti-viral polypeptide or conjugate
thereof to a solid support, such as a filter, can be used in
clinics to remove all or part of the viral content of a
biological solution. For example, filters comprising the
inventive anti-viral agents can be used to treat blood supplies
prior to transfusion to reduce the risk of viral transmission.
Such filters would find particular utility in clinics wherein
risk of viral infection is high. It will be appreciated that
total removal of the viral content of a biological solution is
not required to achieve a beneficial effect. Removal of even a
fraction of virus from a biological solution decreases the risk
of infection of a patient.
[0085] Potential virucides used or being considered for use
against sexual transmission of HIV are very limited; present
agents in this category include, for example, nonoxynol-9 (Bird,
AIDS, 5: 791-796 (1991)), gossypol and derivatives (Polsky et
al., Contraception, 39: 579-587 (1989); Lin, Antimicrob. Agents
Chemother, 33: 2149-2151 (1989); and Royer, Pharmacol. Res, 24:
407-412 (1991)), and gramicidin (Bourinbair, Life
Sci./Pharmacol. Lett, 54: PL5-9 (1994); and Bourinbair et al.,
Contraception, 49: 131-137 (1994)). The method of prevention of
sexual transmission of viral infection, e.g., HIV infection, in
accordance with the invention comprises vaginal, rectal, oral,
penile or other topical treatment with an anti-viral effective
amount of a griffithsin and/or griffithsin conjugate, alone or
in combination with another anti-viral compound as described
herein.
[0086] In a novel approach to anti-HIV prophylaxis pursued under
auspices of the U.S. National Institute of Allergy and
Infectious Diseases (NIAID) (e.g., as conveyed by Painter, USA
Today, Feb. 13, 1996), vaginal suppository instillation of live
cultures of lactobacilli was being evaluated in a 900-woman
study. This study was based especially upon observations of
anti-HIV effects of certain H2O2-producing lactobacilli in vitro
(e.g., see published abstract by Hilier, from NIAID-sponsored
Conference on "Advances in AIDS Vaccine Development," Bethesda,
Md., Feb. 11-15, 1996). Lactobacilli readily populate the
vagina, and indeed are a predominant bacterial population in
most healthy women (Redondo-Lopez et al., Rev. Infect. Dis., 12:
856-872 (1990); Reid et al., Clin. Microbiol. Rev., 3: 335-344
(1990); Bruce and Reid, Can. J. Microbiol., 34: 339-343 (1988);
Reu et al., J. Infect. Dis., 171: 1237-1243 (1995); Hilier et
al., Clin. Infect. Dis., 16 (Suppl 4): S273-S281; and Agnew et
al., Sex. Transm. Dis., 22: 269-273 (1995)). Lactobacilli are
also prominent, nonpathogenic inhabitants of other body cavities
such as the mouth, nasopharynx, upper and lower gastrointestinal
tracts, and rectum.
[0087] It is well-established that lactobacilli can be readily
transduced using available genetic engineering techniques to
incorporate a desired foreign DNA coding sequence, and that such
lactobacilli can be made to express a corresponding desired
foreign protein (see, e.g., Hols et al., Appl. and Environ.
Microbiol., 60: 1401-1413 (1994)). Therefore, within the context
of the present disclosure, it will be appreciated by one skilled
in the art that viable host cells containing a DNA sequence or
vector of the invention, and expressing a polypeptide or fusion
protein of the invention, can be used directly as the delivery
vehicle for a griffithsin or fusion protein thereof to the
desired site(s) in vivo. Preferred host cells for such delivery
of griffithsins or fusion proteins thereof directly to desired
site(s), such as, for example, to a selected body cavity, can
comprise bacteria or yeast. More specifically, such host cells
can comprise suitably engineered strain(s) of lactobacilli,
enterococci, or other common bacteria, such as E. coli, normal
strains of which are known to commonly populate body cavities.
More specifically yet, such host cells can comprise one or more
selected nonpathogenic strains of lactobacilli, such as those
described by Andreu et al. ((1995), supra), especially those
having high adherence properties to epithelial cells, such as,
for example, adherence to vaginal epithelial cells, and suitably
transformed using the DNA sequences of the present invention.
[0088] As reviewed by McGroarty (FEMS Immunol. Med. Microbiol.,
6: 251-264 (1993)) the "probiotic" or direct therapeutic
application of live bacteria, particularly bacteria that occur
normally in nature, more particularly lactobacilli, for
treatment or prophylaxis against pathogenic bacterial or yeast
infections of the urogenital tract, in particular the female
urogenital tract, is a well-established concept. Recently, the
use of a conventional probiotic strategy, in particular the use
of live lactobacilli, to inhibit sexual transmission of HIV has
been suggested, based specifically upon the normal, endogenous
production of virucidal levels of H2O2 and/or lactic acid and/or
other potentially virucidal substances by certain normal strains
of lactobacilli (e.g., Hilier (1996), supra). However, the
inventive use of non-mammalian cells, particularly bacteria,
more particularly lactobacilli, specifically engineered with a
foreign gene, more specifically a griffithsin gene, to express
an anti-viral substance, more specifically a protein, and even
more specifically a griffithsin, is heretofore unprecedented as
a method of treatment of an animal, specifically a human, to
prevent infection by a virus, specifically a retrovirus, more
specifically HIV-1 or HIV-2.
[0089] Elmer et al. (JAMA, 275: 870-876 (1996)) have recently
speculated that "genetic engineering offers the possibility of
using microbes to deliver specific actions or products to the
colon or other mucosal surfaces . . . other fertile areas for
future study include defining the mechanisms of action of
various biotherapeutic agents with the possibility of applying
genetic engineering to enhance activities." Elmer et al.
((1996), supra) further point out that the terms "probiotic" and
"biotherapeutic agent" have been used in the literature to
describe microorganisms that have antagonistic activity toward
pathogens in vivo; those authors more specifically prefer the
term "biotherapeutic agent" to denote "microorganisms having
specific therapeutic properties."
[0090] In view of the present disclosure, one skilled in the art
will appreciate that the invention teaches an entirely novel
type of "probiotic" or "biotherapeutic" treatment using
specifically engineered strains of microorganisms provided
herein which do not occur in nature. Nonetheless, available
teachings concerning selection of optimal microbial strains, in
particular bacterial strains, for conventional probiotic or
biotherapeutic applications can be employed in the context of
the invention. For example, selection of optimal lactobacillus
strains for genetic engineering, transformation, direct
expression of griffithsins or conjugates thereof, and direct
probiotic or biotherapeutic applications, to treat or prevent
viral (e.g., HIV) infection, can be based upon the same or
similar criteria, such as those described by Elmer et al.
((1996), supra), typically used to select normal, endogenous or
"nonengineered" bacterial strains for conventional probiotic or
biotherapeutic therapy. Furthermore, the recommendations and
characteristics taught by McGroarty, particularly for selection
of optimal lactobacillus strains for conventional probiotic use
against female urogenital infections, are pertinent to the
present invention: " . . . lactobacilli chosen for incorporation
into probiotic preparations should be easy and, if possible,
inexpensive to cultivate . . . strains should be stable, retain
viability following freeze-drying and, of course, be
non-pathogenic to the host . . . it is essential that
lactobacilli chosen for use in probiotic preparations should
adhere well to the vaginal epithelium . . . ideally,
artificially implanted lactobacilli should adhere to the vaginal
epithelium, integrate with the indigenous microorganisms
present, and proliferate" (McGroarty (1993), supra). While
McGroarty's teachings specifically address selections of
"normal" lactobacillus strains for probiotic uses against
pathogenic bacterial or yeast infections of the female
urogenital tract, similar considerations will apply to the
selection of optimal bacterial strains for genetic engineering
and "probiotic" or "biotherapeutic" application against viral
infections as particularly encompassed by the present invention.
[0091] Accordingly, the method of the invention for the
prevention of sexual transmission of viral infection, e.g., HIV
infection, comprises vaginal, rectal, oral, penile, or other
topical, insertional, or instillational treatment with an
anti-viral effective amount of a griffithsin, a griffithsin
conjugate or fusion protein, a matrix-anchored griffithsin or
conjugate or fusion protein thereof, and/or viable host cells
transformed to express a griffithsin or conjugate or fusion
protein thereof, alone or in combination with one or more other
anti-viral compound (e.g., as described above). However,
commensal organisms which produce griffithsin or a fragment,
homolog, or conjugate thereof can inhibit viruses other than
HIV. For example, commensal microorganisms that produce the
inventive polypeptide can be instilled in mucosal tissue at the
site of influenza contact, such as nasal or oral mucosa, to
inhibit influenza infection of a host.
[0092] Compositions for use in the prophylactic or therapeutic
treatment methods of the invention comprise one or more
griffithsin(s) or conjugate(s) or fusion protein(s) thereof,
either one of which can be matrix-anchored, and desirably a
carrier therefor, such as a pharmaceutically acceptable carrier.
Pharmaceutically acceptable carriers are well-known to those who
are skilled in the art, as are suitable methods of
administration. The choice of carrier will be determined in part
by the particular griffithsin or conjugate or fusion protein
thereof, as well as by the particular method used to administer
the composition.
[0093] One skilled in the art will appreciate that various
routes of administering a drug are available, and, although more
than one route can be used to administer a particular drug, a
particular route can provide a more immediate and more effective
reaction than another route. For example, the anti-viral agent
of the invention can be inhaled in methods of prophylactically
treating a subject for influenza infection. Delivery of the
anti-viral agent to a location of initial viral contact, such as
the nose or mouth, blocks the onset of infection. The anti-viral
agent can be administered via subcutaneous injection.
Alternatively, in acute or critical medical situations, the
anti-viral agent can be administered intravenously. In many
cases of infection, a patient generates an immune response to a
virus. However, the effects of the viral infection so severely
compromise the health of the patient that an effective immune
response is not reached prior to death. Administration of the
anti-viral agent can prolong the life of the patient until a
patient's natural immune defense clears the virus. Furthermore,
one skilled in the art will appreciate that the particular
pharmaceutical carrier employed will depend, in part, upon the
particular griffithsin or conjugate or fusion protein thereof
employed, and the chosen route of administration. Accordingly,
there is a wide variety of suitable formulations of the
composition of the invention.
[0094] Formulations suitable for oral administration can consist
of liquid solutions, such as an effective amount of the compound
dissolved in diluents, such as water, saline, or fruit juice;
capsules, sachets or tablets, each containing a predetermined
amount of the active ingredient, as solid, granules or
freeze-dried cells; solutions or suspensions in an aqueous
liquid; and oil-in-water emulsions or water-in-oil emulsions.
Tablet forms can include one or more of lactose, mannitol, corn
starch, potato starch, microcrystalline cellulose, acacia,
gelatin, colloidal silicon dioxide, croscarmellose sodium, talc,
magnesium stearate, stearic acid, and other excipients,
colorants, diluents, buffering agents, moistening agents,
preservatives, flavoring agents, and pharmacologically
compatible carriers. Suitable formulations for oral delivery can
also be incorporated into synthetic and natural polymeric
microspheres, or other means to protect the agents of the
present invention from degradation within the gastrointestinal
tract (see, for example, Wallace et al., Science, 260: 912-915
(1993)).
[0095] The anti-viral agent of the invention (e.g., griffithsin
or conjugates thereof), alone or in combination with other
anti-viral compounds, can be made into aerosol formulations or
microparticulate powder formulations to be administered via
inhalation. These aerosol formulations can be placed into
pressurized acceptable propellants, such as
dichlorodifluoromethane, propane, nitrogen, and the like.
[0096] The anti-viral agent of the invention (e.g., griffithsin
or conjugates thereof), alone or in combinations with other
anti-viral compounds or absorption modulators, can be made into
suitable formulations for transdermal application and
absorption, such as a patch (Wallace et al. (1993), supra).
Transdermal electroporation or iontophoresis also can be used to
promote and/or control the systemic delivery of the compounds
and/or compositions of the present invention through the skin
(e.g., see Theiss et al., Meth. Find. Exp. Clin. Pharmacol., 13:
353-359 (1991)).
[0097] Formulations suitable for topical administration include
lozenges comprising the active ingredient in a flavor, usually
sucrose and acacia or tragacanth; pastilles comprising the
active ingredient in an inert base, such as gelatin and
glycerin, or sucrose and acacia; and mouthwashes comprising the
active ingredient in a suitable liquid carrier; as well as
creams, emulsions, gels and the like containing, in addition to
the active ingredient, such as, for example, freeze-dried
lactobacilli or live lactobacillus cultures genetically
engineered to directly produce a griffithsin or conjugate or
fusion protein thereof of the present invention, such carriers
as are known in the art. Topical administration is preferred for
the prophylactic and therapeutic treatment of influenza viral
infection, such as through the use of an inhaler, for example.
[0098] Formulations for rectal administration can be presented
as a suppository with a suitable base comprising, for example,
cocoa butter or a salicylate. Formulations suitable for vaginal
administration can be presented as pessaries, tampons, creams,
gels, pastes, foams, or spray formulas containing, in addition
to the active ingredient, such as, for example, freeze-dried
lactobacilli or live lactobacillus cultures genetically
engineered to directly produce a griffithsin or conjugate or
fusion protein thereof of the present invention, such carriers
as are known in the art to be appropriate. Similarly, the active
ingredient can be combined with a lubricant as a coating on a
condom. Indeed, preferably, the active ingredient is applied to
any contraceptive device, including, but not limited to, a
condom, a diaphragm, a cervical cap, a vaginal ring, and a
sponge.
[0099] Formulations suitable for parenteral administration
include aqueous and non-aqueous, isotonic sterile injection
solutions, which can contain anti-oxidants, buffers,
bacteriostats, and solutes that render the formulation isotonic
with the blood of the intended recipient, and aqueous and
non-aqueous sterile suspensions that can include suspending
agents, solubilizers, thickening agents, stabilizers, and
preservatives. The formulations can be presented in unit-dose or
multi-dose sealed containers, such as ampules and vials, and can
be stored in a freeze-dried (lyophilized) condition requiring
only the addition of the sterile liquid carrier, for example,
water, for injections, immediately prior to use. Extemporaneous
injection solutions and suspensions can be prepared from sterile
powders, granules, and tablets of the kind previously described.
[0100] Formulations comprising a griffithsin or griffithsin
conjugate suitable for virucidal (e.g., HIV) sterilization of
inanimate objects, such as medical supplies or equipment,
laboratory equipment and supplies, instruments, devices, and the
like, can, for example, be selected or adapted as appropriate,
by one skilled in the art, from any of the aforementioned
compositions or formulations. Preferably, the griffithsin is
produced by recombinant DNA technology. The griffithsin
conjugate can be produced by recombinant DNA technology or by
chemical coupling of a griffithsin with an effector molecule as
described above. Similarly, formulations suitable for ex vivo
sterilization, inactivation, or removal of virus, such as
infectious virus, from a sample, such as blood, blood products,
sperm, or other bodily products, such as a fluid, cells, a
tissue or an organ, or any other solution, suspension, emulsion,
vaccine formulation (such as in the removal of infectious
virus), or any other material which can be administered to a
patient in a medical procedure, can be selected or adapted as
appropriate by one skilled in the art, from any of the
aforementioned compositions or formulations. However, suitable
formulations for ex vivo sterilization or inactivation or
removal of virus from a sample or on an inanimate object are by
no means limited to any of the aforementioned formulations or
compositions. For example, such formulations or compositions can
comprise a functional griffithsin, such as that which is encoded
by SEQ ID NO: 3, or anti-viral fragment thereof, such as a
fragment comprising at least eight contiguous amino acids of SEQ
ID NO: 3, wherein the at least eight contiguous amino acids bind
to a virus, or a conjugate or fusion protein of either of the
foregoing, attached to a solid support matrix, to facilitate
contacting or binding infectious virus in a sample or removing
infectious virus from a sample as described above, e.g., a
bodily product such as a fluid, cells, a tissue or an organ from
an organism, in particular a mammal, such as a human, including,
for example, blood, a component of blood (e.g., plasma, blood
cells, and the like), or sperm. Preferably, the anti-viral
polypeptide comprises SEQ ID NO: 3. Also preferably, the at
least eight contiguous amino acids bind gp120 of HIV, in
particular infectious HIV. As a more specific example, such a
formulation or composition can comprise a functional
griffithsin, or conjugate or fusion protein thereof, attached to
(e.g., coupled to or immobilized on) a solid support matrix
comprising magnetic beads, to facilitate contacting, binding and
removal of infectious virus, and to enable magnet-assisted
removal of the virus from a sample as described above, e.g., a
bodily product such as a fluid, cells, a tissue or an organ,
e.g., blood, a component of blood, or sperm. Alternatively, and
also preferably, the solid support matrix comprises a
contraceptive device, such as a condom, a diaphragm, a cervical
cap, a vaginal ring, or a sponge. The anti-viral agent also can
be encapsulated or dispersed within a solid matrix, such as a
vaginal ring or sponge. Methods for encapsulating
biotherapeutics into, for example, biocompatible sustained
release devices, are known in the art.
[0101] As an even more specific illustration, such a composition
(e.g., for ex vivo) can comprise a functional (e.g.,
gp120-binding, HIV-inactivating) griffithsin, or conjugate or
fusion protein thereof, attached to a solid support matrix, such
as magnetic beads or a flow-through matrix, by means of an
anti-griffithsin antibody or at least one effector component,
which can be the same or different, such as polyethylene glycol,
albumin, or dextran. The conjugate can further comprise at least
one effector component, which can be the same or different,
selected from the group consisting of, for example, an
immunological reagent and a toxin. A flow-through matrix would
comprise, for instance, a configuration similar to an affinity
column. The griffithsin can be covalently coupled to a solid
support matrix via an anti-griffithsin antibody, described
below. Methods of attaching an antibody to a solid support
matrix are well-known in the art (see, for example, Harlow and
Lane. Antibodies: A Laboratory Manual, Cold Springs Harbor
Laboratory: Cold Spring Harbor, N.Y. (1988)). Alternatively, the
solid support matrix, such as magnetic beads, can be coated with
streptavidin, in which case the griffithsin or fragment thereof
(which comprises at least eight contiguous amino acids of SEQ ID
NO: 3 or SEQ ID NO: 2), or a conjugate or fusion protein of
either one, is biotinylated. The at least eight contiguous amino
acids of SEQ ID NO: 2 desirably have anti-viral activity and
preferably bind gp120 of HIV, which preferably is infectious.
Preferably, the anti-viral polypeptide comprises SEQ ID NO: 3 or
SEQ ID NO: 2. Such a composition can be prepared, for example,
by biotinylating the griffithsin, or conjugate or fusion protein
thereof, and then contacting the biotinylated protein or peptide
with a (commercially available) solid support matrix, such as
magnetic beads, coated with streptavidin. The use of
biotinylation as a means to attach a desired biologically active
protein or peptide to a streptavidin-coated support matrix, such
as magnetic beads, is well-known in the art.
[0102] One skilled in the art will appreciate that a suitable or
appropriate formulation can be selected, adapted or developed
based upon the particular application at hand.
[0103] For ex vivo uses, such as virucidal treatments of
inanimate objects or materials, blood or blood products, or
tissues, the amount of griffithsin, conjugate thereof, fusion
protein thereof, or composition of any of the foregoing, to be
employed should be sufficient that any virus or virus-producing
cells present will be rendered noninfectious or will be
destroyed. For example, for HIV, this would require that the
virus and/or the virus-producing cells be exposed to
concentrations of griffithsin in the range of 0.1-1000 nM.
Similar considerations apply to in vivo applications. Therefore,
the designation of "anti-viral effective amount" is used
generally to describe the amount of a particular griffithsin,
conjugate, fusion protein, or composition thereof required for
anti-viral efficacy in any given application.
[0104] In view of the above, the invention also provides a
method of inhibiting prophylactically or therapeutically a viral
infection of a host in which an anti-viral effective amount of
an above-described anti-viral polypeptide, conjugate, or fusion
protein is administered to the host. Upon administration of the
anti-viral effective amount of the anti-viral polypeptide,
conjugate, or fusion protein, the viral infection is inhibited.
[0105] The invention additionally provides a method of
prophylactically or therapeutically inhibiting a viral infection
of a host in which an anti-viral effective amount of a
composition comprising an isolated and purified anti-viral
polypeptide, or anti-viral polypeptide conjugate or fusion
protein, either one of which comprises at least eight contiguous
amino acids of SEQ ID NO: 3 having anti-viral activity, attached
to or encapsulated within a solid support matrix is administered
to the host. By "therapeutically" is meant that the host already
has been infected with the virus. By "prophylactically" is meant
that the host has not yet been infected with the virus but is at
risk of being infected with the virus. Prophylactic treatment is
intended to encompass any degree of inhibition of viral
infection, including, but not limited to, complete inhibition,
as one of ordinary skill in the art will readily appreciate that
any degree in inhibition of viral infection is advantageous.
Preferably, the inventive active agent is administered before
viral infection or immediately upon determination of viral
infection and is continuously administered until the virus is
undetectable. The method optionally further comprises the prior,
simultaneous or subsequent administration, by the same route or
a different route, of an antiviral agent or another agent that
is efficacious in inhibiting the viral infection. Upon
administration of the anti-viral effective amount of the
composition, the viral infection is inhibited. Preferably, the
solid support matrix is a contraceptive device, such as a
condom, diaphragm, cervical cap, vaginal ring, or sponge. In an
alternative embodiment, a solid support matrix can be surgically
implanted and later removed.
[0106] For in vivo uses, the dose of a griffithsin, or conjugate
or composition thereof, administered to an animal, particularly
a human, in the context of the invention should be sufficient to
effect a prophylactic or therapeutic response in the individual
over a reasonable time frame. The dose used to achieve a desired
anti-viral concentration in vivo (e.g., 0.1-1000 nM) will be
determined by the potency of the particular griffithsin or
conjugate employed, the severity of the disease state of
infected individuals, as well as, in the case of systemic
administration, the body weight and age of the infected
individual. The size of the dose also will be determined by the
existence of any adverse side effects that may accompany the
particular griffithsin, or conjugate or composition thereof,
employed. It is always desirable, whenever possible, to keep
adverse side effects to a minimum.
[0107] The invention also provides a method of removing virus,
such as infectious virus, from a sample. The method comprises
contacting the sample with a composition comprising an isolated
and purified anti-viral polypeptide or conjugate or fusion
protein thereof, comprising at least eight contiguous amino
acids of SEQ ID NO: 3 (or SEQ ID NO: 2). The at least eight
contiguous amino acids desirably have anti-viral activity and
bind to the virus and the anti-viral polypeptide (or conjugate
or fusion protein of either of the foregoing) is attached to a
solid support matrix, such as a magnetic bead. "Attached" is
used herein to refer to attachment to (or coupling to) and
immobilization in or on a solid support matrix. While any means
of attachment can be used, preferably, attachment is by covalent
bonds. The method further comprises separating the sample and
the composition by any suitable means, whereupon the virus, such
as infectious virus, is removed from the sample. Preferably, the
anti-viral polypeptide comprises SEQ ID NO: 3 (or SEQ ID NO: 2).
In one embodiment, the anti-viral polypeptide is conjugated with
an anti-griffithsin antibody or at least one effector component,
which can be the same or different, selected from polyethylene
glycol, dextran and albumin, in which case the anti-viral
polypeptide is desirably attached to the solid support matrix
through at least one effector component. The anti-viral
polypeptide can be further conjugated with at least one effector
component, which can be the same or different, selected from the
group consisting of an immunological reagent and a toxin. In
another embodiment, the solid support matrix is coated with
streptavidin and the anti-viral polypeptide is biotinylated.
Through biotin, the biotinylated anti-viral polypeptide is
attached to the streptavidin-coated solid support matrix. Other
types of means, as are known in the art, can be used to attach a
functional griffithsin (i.e., an anti-viral polypeptide or
conjugate as described above) to a solid support matrix, such as
a magnetic bead, in which case contact with a magnet is used to
separate the sample and the composition. Similarly, other types
of solid support matrices can be used, such as a matrix
comprising a porous surface or membrane, over or through which a
sample is flowed or percolated, thereby selectively entrapping
or removing infectious virus from the sample. The choice of
solid support matrix, means of attachment of the functional
griffithsin to the solid support matrix, and means of separating
the sample and the matrix-anchored griffithsin will depend, in
part, on the sample (e.g., fluid vs. tissue) and the virus to be
removed. It is expected that the use of a selected coupling
molecule can confer certain desired properties to a matrix,
comprising a functional griffithsin coupled therewith, that may
have particularly advantageous properties in a given situation.
Preferably, the sample is blood, a component of blood, sperm,
cells, tissue or an organ. Also, preferably the sample is a
vaccine formulation, in which case the virus that is removed is
infectious, such as HIV, although HIV, in particular infectious
HIV, can be removed from other samples in accordance with this
method.
[0108] For instance, the skilled practitioner might select a
poly(ethylene glycol) molecule for attaching a functional
griffithsin to a solid support matrix, thereby to provide a
matrix-anchored griffithsin, wherein the griffithsin is attached
to the matrix by a longer "tether" than would be feasible or
possible for other attachment methods, such as
biotinylation/streptavidin coupling. A griffithsin coupled by a
poly(ethylene glycol) "tether" to a solid support matrix (such
as magnetic beads, porous surface or membrane, and the like) can
permit optimal exposure of a binding surface, epitope,
hydrophobic or electrophilic focus, and/or the like, on a
functional griffithsin in a manner that, in a given situation
and/or for a particular virus, facilitates the binding and/or
inactivation of the virus. A preferred solid support matrix is a
magnetic bead such that separation of the sample and the
composition is effected by a magnet. In a preferred embodiment
of the method, the at least eight contiguous amino acids bind
gp120 of HIV and HIV is removed from the sample.
[0109] Similarly, other types of solid support matrices can be
used, such as a matrix comprising a porous surface or membrane,
over or through which a sample is flowed or percolated, thereby
selectively inhibiting infectious virus (e.g., HIV or influenza)
in the sample. The choice of solid support matrix, means of
attachment of the functional griffithsin to the solid support
matrix, and means of separating the sample and the
matrix-anchored griffithsin will depend, in part, on the sample
(e.g., fluid vs. tissue) and the virus to be inhibited. It is
expected that the use of a selected coupling molecule can confer
certain desired properties to a matrix, comprising a functional
griffithsin coupled therewith, that may have particularly
advantageous properties in a given situation.
[0110] The methods described herein also have utility in real
time ex vivo inhibition of virus or virus infected cells in a
bodily fluid, such as blood, e.g., in the treatment of viral
infection, or in the inhibition of virus in blood or a component
of blood, e.g., for transfusion, in the inhibition or prevention
of viral infection. Such methods also have potential utility in
dialysis, such as kidney dialysis, and in inhibiting virus in
sperm obtained from a donor for in vitro and in vivo
fertilization. The methods also have applicability in the
context of tissue and organ transplantations.
[0111] In summary, a griffithsin attached to a solid support
matrix, such as a magnetic bead, can be used to remove virus, in
particular infectious virus, including immunodeficiency virus,
such as HIV, e.g., HIV-1 or HIV-2, from a sample, such as a
sample comprising both infectious and noninfectious virus. The
inventive method also can be used to remove viral glycoprotein
presenting cells, e.g., infected cells that have, for example,
gp120 on their surfaces, from a sample.
[0112] The invention, therefore, further provides a composition
comprising naturally-occurring, non-infectious virus, such as a
composition produced as described above. The composition can
further comprise a carrier, such as a biologically or
pharmaceutically acceptable carrier, and an immuno-adjuvant.
Preferably, the noninfectious virus is an influenza or an
immunodeficiency virus, such as HIV, e.g., HIV-1 or HIV-2.
Alternatively, and also preferably, the noninfectious virus is
FIV. A composition comprising only naturally-occurring,
non-infectious virus has many applications in research and the
prophylactic treatment of a viral infection. In terms of
prophylactic treatment of a viral infection, the skilled artisan
will appreciate the need to eliminate completely all infectious
virus from the composition. If desired, further treatment of the
composition comprising non-infectious particles with
virus-inactivating chemicals, such as imines or psoralens,
and/or pressure or heat inactivation, will further the
non-infectious nature of the composition. For example, an immune
response-inducing amount of the inventive composition can be
administered to an animal at risk for a viral infection in order
to induce an immune response. The skilled artisan will
appreciate that such a composition is a significant improvement
over previously disclosed compositions in that the virus is
non-infectious and naturally-occurring. Thus, there is no risk
of inadvertent infection, greater doses can be administered in
comparison to compositions comprising infectious viral
particles, and the subsequent immune response will assuredly be
directed to antigens present on naturally-occurring virus. The
composition comprising naturally-occurring, non-infectious virus
can be administered in any manner appropriate to induce an
immune response. Preferably, the virus is administered, for
example, intramuscularly, mucosally, intravenously,
subcutaneously, or topically. Preferably, the composition
comprises naturally-occurring, non-infectious human
immunodeficiency virus comprising gp120.
[0113] The composition comprising naturally-occurring,
non-infectious virus can be combined with various carriers,
adjuvants, diluents or other anti-viral therapeutics, if
desired. Appropriate carriers include, for example, ovalbumin,
albumin, globulins, hemocyanins, and the like. Adjuvants or
immuno-adjuvants are incorporated in most cases to stimulate
further the immune system. Any physiologically appropriate
adjuvant can be used. Suitable adjuvants for inclusion in the
inventive composition include, for example, aluminum hydroxide,
beryllium sulfate, silica, kaolin, carbon, bacterial endotoxin,
saponin, and the like.
[0114] Thus, the invention also provides a method of inducing an
immune response to a virus in an animal. The method comprises
administering to the animal an immune response-inducing amount
of a composition comprising naturally-occurring, non-infectious
virus as described above.
[0115] The appropriate dose of a composition comprising
naturally-occurring, non-infectious virus required to induce an
immune response to the virus in an animal is dependent on
numerous factors, such as size of the animal and immune
competency. The amount of composition administered should be
sufficient to induce a humoral and/or cellular immune response.
The amount of non-infectious virus in a particular composition
can be determined using routine methods in the art, such as the
Coulter HIV p24 antigen assay (Coulter Corp., Hialeah, Fla.).
Any suitable dose of a composition comprising non-infectious
virus is appropriate so long as an immune response is induced,
desirably without the appearance of harmful side effects to the
host. In this regard, compositions comprising from about 10<1
>to about 10<5 >particles, preferably from about
10<2 >to about 10<4 >particles, most preferably
about 10<3 >particles, are suitable for inducing an immune
response.
[0116] One of ordinary skill can determine the effectiveness of
the composition to induce an immune response using routine
methods known in the art. Cell-mediated response can be
determined by employing, for example, a virus antigen-stimulated
T-cell proliferation assay. The presence of a humoral immune
response can be determined, for instance, with the Enzyme Linked
Immunosorbent Assay (ELISA). The skilled artisan will appreciate
that there are numerous other suitable assays for evaluating
induction of an immune response. To the extent that a dose is
inadequate to induce an appropriate immune response, "booster"
administrations can subsequently be administered in order to
prompt a more effective immune response.
[0117] In terms of administration of the inventive anti-viral
agents or conjugates thereof, the dosage can be in unit dosage
form, such as a tablet or capsule. The term "unit dosage form"
as used herein refers to physically discrete units suitable as
unitary dosages for human and animal subjects, each unit
containing a predetermined quantity of a griffithsin or
conjugate thereof, alone or in combination with other anti-viral
agents, calculated in an amount sufficient to produce the
desired effect in association with a pharmaceutically acceptable
diluent, carrier, or vehicle.
[0118] The specifications for the unit dosage forms of the
invention depend on the particular griffithsin, or conjugate or
composition thereof, employed and the effect to be achieved, as
well as the pharmacodynamics associated with each griffithsin,
or conjugate or composition thereof, in the host. The dose
administered should be an "anti-viral effective amount" or an
amount necessary to achieve an "effective level" in the
individual patient.
[0119] Since the "effective level" is used as the preferred
endpoint for dosing, the actual dose and schedule can vary,
depending upon interindividual differences in pharmacokinetics,
drug distribution, and metabolism. The "effective level" can be
defined, for example, as the blood or tissue level (e.g.,
0.1-1000 nM) desired in the patient that corresponds to a
concentration of one or more griffithsin or conjugate thereof,
which inhibits a virus, such as HIV, in an assay known to
predict for clinical anti-viral activity of chemical compounds
and biological agents. The "effective level" for agents of the
invention also can vary when the griffithsin, or conjugate or
composition thereof, is used in combination with AZT or other
known anti-viral compounds or combinations thereof.
[0120] One skilled in the art can easily determine the
appropriate dose, schedule, and method of administration for the
exact formulation of the composition being used, in order to
achieve the desired "effective concentration" in the individual
patient. One skilled in the art also can readily determine and
use an appropriate indicator of the "effective concentration" of
the compounds of the invention by a direct (e.g., analytical
chemical analysis) or indirect (e.g., with surrogate indicators
such as p24 or RT) analysis of appropriate patient samples
(e.g., blood and/or tissues).
[0121] In the treatment of some virally infected individuals, it
can be desirable to utilize a "mega-dosing" regimen, wherein a
large dose of the griffithsin or conjugate thereof is
administered, time is allowed for the drug to act, and then a
suitable reagent is administered to the individual to inactivate
the drug.
[0122] The pharmaceutical composition can contain other
pharmaceuticals, in conjunction with the griffithsin or
conjugate thereof, when used to therapeutically treat a viral
infection, such as an influenza infection or an HIV infection
which results in AIDS. Representative examples of these
additional pharmaceuticals include anti-viral compounds,
virucides, immunomodulators, immunostimulants, antibiotics and
absorption enhancers. Exemplary anti-viral compounds include
cyanovirin, AZT, ddI, ddC, gancylclovir, fluorinated
dideoxynucleosides, nonnucleoside analog compounds, such as
nevirapine (Shih et al., PNAS, 88: 9878-9882 (1991)), TIBO
derivatives, such as R82913 (White et al., Anti-viral Res., 16:
257-266 (1991)), BI-RJ-70 (Merigan, Am. J. Med., 90 (Suppl. 4A):
8S-17S (1991)), michellamines (Boyd et al., J. Med. Chem., 37:
1740-1745 (1994)) and calanolides Kashman et al., J. Med. Chem.,
35: 2735-2743 (1992)), nonoxynol-9, gossypol and derivatives,
gramicidin (Bourinbair et al. (1994), supra), neuraminidase
inhibitors, amantadine, enfurtide, and the like. Exemplary
immunomodulators and immunostimulants include various
interleukins, sCD4, cytokines, antibody preparations, blood
transfusions, and cell transfusions. Exemplary antibiotics
include antifungal agents, antibacterial agents, and
anti-Pneutnoeystitis carnii agents. Exemplary absorption
enhancers include bile salts and other surfactants, saponins,
cyclodextrins, and phospholipids (Davis (1992), supra).
[0123] Administration of a griffithsin or conjugate or fusion
protein thereof with other anti-retroviral agents and
particularly with known RT inhibitors, such as ddC, AZT, ddI,
ddA, or other inhibitors that act against other HIV proteins,
such as anti-TAT agents, is expected to inhibit most or all
replicative stages of the viral life cycle. The dosages of ddC
and AZT used in AIDS or ARC patients have been published. A
virustatic range of ddC is generally between 0.05 [mu]M to 1.0
[mu]M. A range of about 0.005-0.25 mg/kg body weight is
virustatic in most patients. The preliminary dose ranges for
oral administration are somewhat broader, for example 0.001 to
0.25 mg/kg given in one or more doses at intervals of 2, 4, 6,
8, 12, etc. hours. Currently, 0.01 mg/kg body weight ddC given
every 8 hrs is preferred. When given in combined therapy, the
other anti-viral compound, for example, can be given at the same
time as the griffithsin or conjugate thereof or the dosing can
be staggered as desired. The two drugs also can be combined in a
composition. Doses of each can be less when used in combination
than when either is used alone.
[0124] It will also be appreciated by one skilled in the art
that a DNA sequence of a griffithsin or conjugate thereof of the
invention can be inserted ex vivo into mammalian cells
previously removed from a given animal, in particular a human,
host. Such cells can be employed to express the corresponding
griffithsin or conjugate or fusion protein in vivo after
reintroduction into the host. Feasibility of such a therapeutic
strategy to deliver a therapeutic amount of an agent in close
proximity to the desired target cells and pathogens, i.e.,
virus, more particularly retrovirus, specifically HIV and its
envelope glycoprotein gp120, has been demonstrated in studies
with cells engineered ex vivo to express sCD4 (Morgan et al.
(1994), supra). It is also possible that, as an alternative to
ex vivo insertion of the DNA sequences of the invention, such
sequences can be inserted into cells directly in vivo, such as
by use of an appropriate viral vector. Such cells transfected in
vivo are expected to produce anti-viral amounts of griffithsin
or a conjugate or fusion protein thereof directly in vivo.
[0125] Given the present disclosure, it will be additionally
appreciated that a DNA sequence corresponding to a griffithsin
or conjugate thereof can be inserted into suitable nonmammalian
host cells, and that such host cells will express therapeutic or
prophylactic amounts of a griffithsin or conjugate or fusion
protein thereof directly in vivo within a desired body
compartment of an animal, in particular a human. Example 5
illustrates the transformation and expression of effective
virucidal amounts of a griffithsin in a non-mammalian cell, more
specifically a bacterial cell. In a preferred embodiment of the
invention, a method of female-controllable prophylaxis against
HIV infection comprises the intravaginal administration and/or
establishment of, in a female human, a persistent intravaginal
population of lactobacilli that have been transformed with a
coding sequence of the invention to produce, over a prolonged
time, effective virucidal levels of a griffithsin or conjugate
thereof, directly on or within the vaginal and/or cervical
and/or uterine mucosa. It is noteworthy that both the World
Health Organization (WHO), as well as the U.S. National
Institute of Allergy and Infectious Diseases, have pointed to
the need for development of female-controlled topical
microbicides, suitable for blocking the transmission of HIV, as
an urgent global priority (Lange et al., Lancet, 341: 1356
(1993); Fauci, NIAID News, Apr. 27, 1995). A composition
comprising the inventive anti-viral agent and a solid-support
matrix is particularly useful in this regard, particularly when
the solid-support matrix is a contraceptive device, such as a
condom, a diaphragm, a cervical cap, a vaginal ring, or a
sponge. In another embodiment, a colony of commensal organisms
transduced with the nucleic acid of the invention and producing
the inventive anti-viral agent is applied to mucosal tissue
associated with the onset of influenza infection, such as
respiratory or oral mucosal.
[0126] The invention also provides antibodies directed to the
polypeptides of the invention. The availability of antibodies to
any given protein is highly advantageous, as it provides the
basis for a wide variety of qualitative and quantitative
analytical methods, separation and purification methods, and
other useful applications directed to the subject polypeptides.
Accordingly, given the present disclosure and the polypeptides
of the invention, it will be readily apparent to one skilled in
the art that antibodies, in particular antibodies specifically
binding to a polypeptide of the invention, can be prepared using
well-established methodologies (e.g., such as the methodologies
described in detail by Harlow and Lane, in Antibodies. A
Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring
Harbor (1988), pp. 1-725). Such antibodies can comprise both
polyclonal and monoclonal antibodies. Furthermore, such
antibodies can be obtained and employed either in solution-phase
or coupled to a desired solid-phase matrix, such as magnetic
beads or a flow through matrix. Having in hand such antibodies
as provided by the invention, one skilled in the art will
further appreciate that such antibodies, in conjunction with
well-established procedures (e.g., such as described by Harlow
and Lane (1988), supra) comprise useful methods for the
detection, quantification, or purification of a griffithsin,
conjugate thereof, or host cell transformed to produce a
griffithsin or conjugate or fusion protein thereof. Example 6
further illustrates an antibody that specifically binds to a
griffithsin. Accordingly, the invention further provides a
composition comprising an anti-griffithsin antibody bound to the
anti-viral agent of the invention, preferably an anti-viral
polypeptide comprising at least eight contiguous amino acids of
SEQ ID NO: 3.
[0127] Matrix-anchored anti-griffithsin antibodies also can be
used in a method to remove virus in a sample. Preferably, the
antibody binds to an epitope of an anti-viral polypeptide of SEQ
ID NO: 2 or SEQ ID NO: 3. Preferably, the matrix is a solid
support matrix, such as a magnetic bead or a flow-through
matrix. If the solid support matrix to which the
anti-griffithsin antibody is attached comprises magnetic beads,
removal of the antibody-griffithsin-virus complex can be readily
accomplished using a magnet.
[0128] In view of the above, the invention provides a method of
removing virus from a sample. The method comprises (a)
contacting the sample with a composition comprising an isolated
and purified anti-viral polypeptide or conjugate or fusion
protein thereof, wherein (i) the anti-viral polypeptide
comprises at least eight contiguous amino acids of SEQ ID NO: 3,
and (ii) the at least eight contiguous amino acids bind to the
virus, and (b) contacting the sample with an anti-griffithsin
antibody attached to a solid support matrix, whereupon the
anti-griffithsin antibody binds to the anti-viral polypeptide or
conjugate or fusion protein thereof to which is bound the virus,
and (c) separating the solid support matrix from the sample,
whereupon the virus is removed from the sample. Preferably, the
anti-viral polypeptide comprises SEQ ID NO: 3. Desirably, the
virus that is removed is infectious, such as HIV. The sample can
be blood, a component of blood, sperm, cells, tissue or an
organ.
[0129] The antibody for use in the aforementioned method is an
antibody that binds to a polypeptide comprising at least eight
contiguous amino acids of SEQ ID NO: 3, and, which polypeptide
can bind to and inactivate a virus. The antibody can be coupled
to the solid support matrix using similar methods and with
similar considerations as described above for attaching a
griffithsin to a solid support matrix. For example, coupling
methods and molecules employed to attach an anti-griffithsin
antibody to a solid support matrix, such as magnetic beads or a
flow-through matrix, can employ biotin/streptavidin coupling or
coupling through molecules, such as polyethylene glycol, albumin
or dextran. Also analogously, it can be shown that, after such
coupling, the matrix-anchored anti-griffithsin antibody retains
its ability to bind to a polypeptide comprising at least eight
contiguous amino acids of SEQ ID NO: 3, which polypeptide can
bind to and inactivate a virus.
[0130] The invention also provides an anti-griffithsin antibody
that is anti-idiotypic in respect to a viral glycoprotein, such
as gp120, i.e., has an internal image of gp120 of a primate
immunodeficiency virus. Preferably, the antibody can compete
with gp120 of a primate immunodeficiency virus for binding to a
griffithsin. In this regard, the primary immunodeficiency virus
preferably is HIV-1 or HIV-2 and the griffithsin preferably
consists essentially of SEQ ID NO: 2 or SEQ ID NO: 3.
Anti-idiotypic antibodies can be generated in accordance with
methods known in the art (see, for example, Benjamin, in
Immunology: a short course, Wiley-Liss, N Y (1996), pp. 436-437;
Kuby, in Immunology, 3rd ed., Freeman, N.Y. (1997), pp. 455-456;
Greenspan et al., FASEB J., 7: 437-443 (1993); and Poskitt,
Vaccine, 9: 792-796 (1991)). Such an anti-idiotypic (in respect
to gp120) anti-griffithsin antibody is useful in a method of
inhibiting infection of an animal with a virus as provided
herein.
[0131] In view of the above, a griffithsin can be administered
to an animal, the animal generates anti-griffithsin antibodies,
among which are antibodies that have an internal image of a
viral glycoprotein, such as gp120. In accordance with well-known
methods, polyclonal or monoclonal antibodies can be obtained,
isolated, and selected. Selection of an anti-griffithsin
antibody that has an internal image of gp120 can be based upon
competition between the anti-griffithsin antibody and gp120 for
binding to a griffithsin, or upon the ability of the
anti-griffithsin antibody to bind to a free griffithsin as
opposed to a griffithsin bound to gp120. Such an
anti-griffithsin antibody can be administered to an animal to
inhibit a viral infection in accordance with methods provided
herein. Although nonhuman anti-idiotypic antibodies, such as an
anti-griffithsin antibody that has an internal image of gp120
and, therefore, is anti-idiotypic to gp120, are proving useful
as vaccine antigens in humans, their favorable properties might,
in certain instances, be further enhanced and/or their adverse
properties further diminished, through "humanization"
strategies, such as those recently reviewed by Vaughan (Nature
Biotech., 16: 535-539 (1998)). Alternatively, a griffithsin can
be directly administered to an animal to inhibit a viral
infection in accordance with methods provided herein such that
the treated animal, itself, generates an anti-griffithsin
antibody that has an internal image of gp120. The production of
anti-idiotypic antibodies, such as anti-griffithsin antibody
that has an internal image of gp120 and, therefore, is
anti-idiotypic to gp120, in an animal to be treated is known as
"anti-idiotype induction therapy," and is described by
Madiyalakan et al. (Hybridoma, 14: 199-203 (1995)), for example.
[0132] In view of the above, the invention enables another
method of inhibiting infection of an animal, such as a mammal,
in particular a human, with a virus. The method comprises
administering to the animal an anti-griffithsin antibody, or a
composition comprising same, in an amount sufficient to induce
in the animal an immune response to the virus, whereupon the
infection of the animal with the virus is inhibited. Preferably,
the anti-griffithsin antibody has an internal image of a viral
glycoprotein, such as gp120 of an immunodeficiency virus with
which the animal can be infected, such as a primate
immunodeficiency virus. Preferably, the antibody can compete
with, for example, gp120 of a primate immunodeficiency virus for
binding to a griffithsin. In this regard, the primate
immunodeficiency virus preferably is HIV-1 or HIV-2 and the
griffithsin preferably consists essentially of SEQ ID NO: 3 or
SEQ ID NO: 2. The method can further comprise the administration
of an immunostimulant.
[0133] Also enabled by the invention is yet another method of
inhibiting infection of an animal, such as a mammal, in
particular a human, with a virus. The method comprises
administering to the animal a griffithsin, which binds a viral
glycoprotein, such as gp120 of an immunodeficiency virus with
which the animal can be infected, in an amount sufficient to
induce in the animal an anti-griffithsin antibody in an amount
sufficient to induce an immune response to a virus sufficient to
inhibit infection of the animal with the virus. Preferably, the
anti-griffithsin antibody has an internal image of gp120 of an
immunodeficiency virus with which the animal can be infected,
such as a primate immunodeficiency virus. Preferably, the
antibody can compete with gp120 of a primate immunodeficiency
virus for binding to a griffithsin. In this regard, the primate
immunodeficiency virus preferably is HIV-1 or HIV-2 and the
griffithsin preferably consists essentially of SEQ ID NO: 2 or
SEQ ID NO: 3.
[0134] With respect to the above methods, sufficient amounts can
be determined in accordance with methods known in the art.
Similarly, the sufficiency of an immune response in the
inhibition of a viral infection in an animal also can be
assessed in accordance with methods known in the art.
[0135] Either one of the above methods can further comprise
concurrent, pre- or post-treatment with an adjuvant to enhance
the immune response, such as the prior, simultaneous or
subsequent administration, by the same or a different route, of
an antiviral agent or another agent that is efficacious in
inducing an immune response to the virus, such as an
immunostimulant. See, for example, Harlow et al. (1988), supra.
[0136] The inventive griffithsins, conjugates, host cells,
antibodies, compositions and methods are further described in
the context of the following examples. These examples serve to
illustrate further the present invention and are not intended to
limit the scope of the invention.
**EXAMPLES****Example 1**
[0137] This example illustrates a method of isolating and
purifying griffithsin from Griffithsin sp. and elucidating the
griffithsin amino acid sequence.
[0138] Anti-HIV bioassay guided fractionation was used to track
the isolation of the griffithsin polypeptide. In brief, the
cellular mass from Griffithsia sp. was harvested by filtration,
freeze-dried, and extracted first with H2O followed by (1:1)
MeOH-CH2Cl2. Individual aliquots of the organic and aqueous
extracts were tested for cytoprotective properties in the NCI
primary anti-HIV screen (Weislow et al. J. Natl. Cancer Inst.,
81: 577-586 (1989)). Only the H2O extract showed anti-HIV
activity.
[0139] A freeze-dried aqueous extract (10 g) was brought to a
concentration of 50 mg/ml by addition of DDH2O and maintained on
ice. Crystalline ammonium sulfate (Sigma, St. Louis, Mo.;
molecular biology grade) was added to the solution such that the
final concentration of the mixture was 75% saturation. The
mixture was allowed to precipitate on ice over night, and was
then centrifuged at 3000 rpm for 50 min. The resulting pellets
were set aside. The supernatant was brought to 1 M ammonium
sulfate followed by another round of precipitation and
centrifugation. The pellets from the second centrifugation were
saved, and the resulting supernatant was filtered using a 0.22
[mu]m filter and subjected to hydrophobic interaction
chromatography. A BioCad workstation (Perseptive Biosystems) was
used for the following column chromatographies. The protein
solution from the centrifugation and filtration steps was
injected onto a Poros PE column (10\*100 mm, Perseptive
Biosystems) pre-equilibrated with a starting buffer of 50 mM
sodium phosphate, 1.5 M ammonium sulfate, pH 7.5. The column was
eluted at a flow rate of 15 ml/min over the following gradient:
(1) 7 column volumes (CV, equal to 7.85 ml) of the starting
buffer; (2) 1.5-0 M ammonium sulfate over 2 CV; (3) 0 M ammonium
sulfate for 15 CV. The eluate was monitored for both
conductivity and absorbance (280 nm). Ammonium sulfate was added
to the void fraction possessing anti-HIV activity to bring the
final concentration to 75% saturation. The mixture was allowed
to precipitate on ice overnight, and was then centrifuged at
3000 rpm for 50 min. DDH2O-resuspended pellets were first
concentrated using a 10 kDa molecular weight limit membrane,
dialyzed against 0.02% sodium azide, and then brought up to a
concentration of 25 mM Tris-HCl, pH 8.5. The resulting protein
solution was injected onto a Poros HQ anion exchange column
(10\*100 mm, Perseptive Biosystems) pre-equilibrated with a
starting buffer of 25 mM Tris-HCl, pH 8.5. The column was eluted
at a flow rate of 15 ml/min using the following gradient: (1) 5
CV of the starting buffer; (2) 0-1 M sodium chloride over 20 CV;
(3) 1 M sodium chloride for 5 CV. The eluate was monitored for
absorbance (280 nm). Active fractions from the HQ column were
concentrated and desalted using a 10 kDa molecular weight limit
membrane and subjected to a Bio-RP C4 reverse phase column
(4.6\*100 mm, Covance, Princeton, N.J.) and eluted at a flow rate
of 4 ml/min using the following gradient: (1) 10 CV of the
starting buffer of 5% acetonitrile in H2O; (2) 5-95%
acetonitrile in H2O over 2.5 CV; (3) 95% acetonitrile in H2O for
5 CV. The eluate was monitored for absorbance (280 nm), and the
active fraction was pooled, lyophilized, and resuspended in
phosphate-buffered saline (PBS), pH 7.4. The protein solution
was injected onto a G3000PW gel permeation column (21.5\*600 mm,
TosoHaas, Montgomeryville, Pa.) and eluted with PBS, pH 7.4, at
a flow rate of 5 ml/min.
[0140] Molecular mass and purity (>99%) of griffithsin were
confirmed by Electrospray ionization mass spectrometry (ESI-MS),
and the protein concentrations were determined by amino acid
analysis. Native molecular weight was determined by calibrating
standard proteins (albumin (68 kDa), cytochrome c (12.5 kDa),
and aprotinin (6.5 kDa)) by their retention time (as measured by
absorbance at 280 nm) and comparing the resulting calibration
curve to the retention time of the active protein. Amino acid
analysis was accomplished using a Beckman Model 6300 Automated
Amino Acid Analyzer according to manufacturer protocols.
N-terminal amino acid sequencing was performed using an Applied
Biosystems Model 4774A Sequencer according to manufacturer
protocols. Matrix-assisted laser desorption ionization-time of
flight mass spectroscopy (MALDI-TOF MS) was performed using a
Kratos Kompact Maldi III instrument (Shimadzu, Columbia, Md.)
operated in a linear mode using sinapinic acid as a matrix and
trypsin as an external standard. ESI-MS was performed with a
JEOL SX102 equipped with an Analytica electrospray source. The
spectrometer was calibrated using a lysozyme standard (molecular
weight=14305.2) prior to each analysis. Samples were injected
into the source in a 1:1 solution of hexafluorosopropanol and 2%
acetic acid. The masses reported were averages calculated from
the various charged states observed.
[0141] Griffithsin was subjected to digestion with cyanogen
bromide (CNBr) and a variety of endoproteinases (Lys-C, Arg-C,
and Asp-N) per manufacturer's instructions. The cleaved peptide
products were purified by reversed-phase HPLC using a gradient
of 0.05% aqueous trifluoroacetic acid for 20 min, then
increasing to 60% acetonitrile in 0.05% aqueous trifluoroacetic
acid over 100 min. Amino acid sequences were determined by
sequential Edman degradation using an Applied Biosystems Model
494 sequencer according to the protocols of the manufacturer,
and the masses of cleaved peptides were analyzed by MALDI-TOF
mass spectrometer. The amino acid sequence of the native
griffithsin polypeptide is set forth as SEQ ID NO: 3.
[0142] In summary, the preliminary analysis of the crude aqueous
extract of algae Griffithsia sp. in the NCI's primary in vitro
anti-HIV screening assay (Weislow et al., supra) identified a
protein that bound soluble gp120. The process described herein
is illustrated in FIG. 1. Anti-HIV bioassay-guided fractionation
of the aqueous resulted in the isolation of griffithsin. The
aqueous extract was subjected to ammonium sulfate precipitation,
hydrophobic interaction chromatography, anion exchange
chromatography, reversed-phase chromatography, and size
exclusion chromatography to produce a homogeneous protein
fraction. SDS-PAGE analysis showed a single protein band with a
relative molecular mass of approximately 13 kDa, named
griffithsin. Purified griffithsin exhibited a single band by
immunoblotting with anti-griffithsin polyclonal antibodies. The
amino acid sequence of the purified griffithsin was established
by N-terminal Edman degradation of the intact protein and by
N-terminal sequencing of peptide fragments cleaved by CNBr and a
variety of endopeptidases (Lys-C, Arg-C, and Asp-N) followed by
reversed phase purification and MALDI-TOF mass spectrometric
analysis. The entire 121 amino acid sequence was established
except for a single amino acid at position 31, which does not
match any of the common amino acids. Electrospray ionization
mass spectrometric analysis of isolated griffithsin showed a
molecular ion with m/z 12,770.05, and the calculated value for
the deduced amino acid sequence without amino acid at position
31 was m/z 12619.00. It was deduced that the molecular mass of
the amino acid at position 31 was 151.05. The amino acid
analysis of griffithsin also agreed with the deduced primary
sequence. These data fully support the proposed primary amino
acid sequence of griffithsin. A search of the BLAST database
(Altschul et al., Nucleic Acids Res, 25 (17), 3389-3402 (1997))
for identification of protein sequence similarities did not
reveal any homologies of greater than eight contiguous amino
acids nor >30% total sequence homology between griffithsin
and any amino acid sequences of known proteins or transcription
products of known nucleotide sequences, including the anti-HIV
proteins cyanovirin-N and scytovirin.
**Example 2**
[0143] This example demonstrates the synthesis of griffithsin
genes. The methods described herein are illustrated in FIG. 2.
[0144] The chemically deduced amino acid sequence of griffithsin
was back-translated to elucidate the corresponding DNA coding
sequence. Since amino acid residue 31 of native griffithsin did
not appear to be one of the twenty common amino acids, alanine
was substituted in this position (SEQ ID NO: 2). In order to
facilitate initial production and purification of recombinant
griffithsin, a commercial expression vector pET-26b(+), from
Novagen, Inc., Madison, Wis., for which reagents were available
for affinity purification and detection, was selected.
Appropriate restriction sites for ligation to pET-26b(+), and a
stop codon, were included in the DNA sequence. SEQ ID NO: 1 is
an example of a DNA sequence encoding a synthetic griffithsin
gene. A flowchart illustrating a method of synthesizing of a
griffithsin gene is shown in FIG. 2.
[0145] A griffithsin-encoding DNA sequence was synthesized as 13
overlapping, complementary oligonucleotides and assembled to
form the double-stranded coding sequence. Oligonucleotide
elements of the synthetic DNA coding sequence were synthesized
using a nucleic acid synthesizer (model 394, Applied Biosystems
Inc., Foster City, Calif.). The purified 13 oligonucleotides
were individually treated with T4 polynucleotide kinase, and 1
nM quantities of each were pooled and boiled for 10 minutes to
ensure denaturation. The temperature of the mixture was then
reduced to 70[deg.] C. for annealing of the complementary
strands for 15 minutes, and further reduced to 60[deg.] C. for
15 minutes. The reaction was cooled on ice and T4 DNA ligase
(2,000 units) additional ligase buffer was added to the
reaction. Ligation of the oligonucleotides was performed with T4
DNA ligase overnight at 16[deg.] C. The resulting DNA was
recovered and purified from the reaction buffer by
phenol:chloroform extraction, ethanol precipitation, and further
washing with ethanol.
[0146] The purified, double-stranded synthetic DNA was then used
as a template in a polymerase chain reaction (PCR). One [mu]l of
the DNA solution obtained after purification of the ligation
reaction mixture was used as a template. Thermal cycling was
performed using a Perkin-Elmer instrument. "Pfu" thermostable
DNA polymerase, restriction enzymes, T4 DNA ligase, and
polynucleotide kinase were obtained from Stratagene, La Jolla,
Calif. Pfu polymerase was selected for this application because
of its claimed superiority in fidelity compared to the usual Taq
enzyme. The PCR reaction product was run on a 2% agarose gel in
TAE buffer. The 465 base pair DNA construct was cut from the gel
and purified. The purified DNA, which was digested with Nde I
and Xho I restriction enzymes, was then ligated into the
multicloning site of the pet-26b(+) vector.
[0147] E. coli were transfected with the generated
pET-26b(+)-construct, and recombinant clones were identified by
analysis of restriction digests of plasmid DNA. Sequence
analysis of one of these selected clones indicated that three
bases deviated from the intended coding sequence. These
"mutations," which presumably arose during the PCR amplification
of the synthetic template, were corrected by a site-directed
mutagenesis kit from Stratagene, La Jolla, Calif. The repair was
confirmed by DNA sequence analysis.
[0148] For preparation of a DNA sequence encoding a griffithsin
polypeptide tagged with a penta-His peptide at the C-terminal
end of griffithsin (e.g., SEQ ID NO: 4), the aforementioned
recombinant griffithsin construct was subjected to site-directed
mutagenesis to eliminate stop codons located between the
griffithsin coding sequence and the penta-His peptide coding
sequence using a site-directed mutagenesis kit from Stratagene,
La Jolla, Calif. A pair of mutagenic oligonucleotide primers
were synthesized, which included portions of the codons encoding
the griffithsin polypeptide and penta-His peptide, but lacked
the stop codons. Annealing of these mutagenic primers with the
template DNA and extension by DNA polymerase resulted in the
generation of a DNA construct encoding a fusion protein
comprising the griffithsin amino acid sequence linked to a
penta-His peptide tag. DNA sequencing verified the presence of
the intended sequence.
**Example 3**
[0149] This example demonstrates the expression of an N-terminal
His-tagged-griffithsin gene.
[0150] A recombinant griffithsin protein and a C-terminal,
His-tagged griffithsin protein encoded by the nucleic acids of
Example 2 did not efficiently translocate to the periplasmic
fraction of E. coli following protein expression. In addition,
the majority of the produced proteins accumulated in the
inclusion bodies of E. coli without the cleavage of a pelB
signal sequence located at the N-terminus of the griffithsin
protein. Thus, steps were taken to express griffithsin in the
cytosolic fraction of E. coli.
[0151] The pET-26b(+)-griffithsin DNA construct was used as a
template PCR using a pair of appropriate primers. The PCR
product was designed to have a "penta-His" peptide and thrombin
recognition site at the N-terminal end of the griffithsin
polypeptide, providing for production of a N-terminal,
His-tagged-griffithsin fusion protein. The PCR reaction product
was purified from an agarose gel. The purified DNA, which was
digested with Nco I and Xho I restriction enzymes, was ligated
into the expression vector pET-28a(+) vector (Novagen, Inc.,
Madison, Wis.).
[0152] E. coli (strain BL21 (DE3)) were transfected with the
pET-28a(+) vector containing the nucleic acid coding sequence
for the His-tagged-griffithsin fusion protein (see SEQ ID NO:
4). Selected clones were seeded into small-scale shake flasks
containing LB growth medium with 30 [mu]g/ml kanamycin and
expanded by incubation at 37[deg.] C. Larger-scale Erlenmeyer
flasks (0.5-3.0 liters) were then seeded. The culture was
allowed to grow to a density of 0.5-0.7 OD600 units. The
expression of the His-tagged-griffithsin fusion protein was
induced by adding IPTG to a final concentration of 1 mM and
continuing incubation at 37[deg.] C. for 3-6 hrs. Bacteria were
harvested by centrifugation, and the soluble fraction was
obtained using BugBuster(TM) reagent and Benzonase nuclease
(Novagen, Inc., Madison, Wis.). Crude soluble fractions showed
both anti-HIV activity and presence of a His-tagged-griffithsin
fusion protein by Western-blotting. In addition, the
His-tagged-griffithsin protein accumulated in the inclusion
bodies of E. coli. A flowchart illustrating a method of
expressing and purifying recombinant His-tagged-griffithsin is
shown in FIG. 3.
[0153] The purity (~98%) of recombinant His-tagged griffithsin
was confirmed by SDS-PAGE on 16% Tricine gel stained by
Coomassie Blue staining. The protein showed the expected
molecular mass for griffithsin (i.e., 14.6 kDa). Protein
concentrations were determined based on extinction coefficient
at 280 nm of the protein. Approximately 1.6 mg of recombinant
His-tagged griffithsin was purified from 1 L of E. coli culture.
The purified protein demonstrated gp120-binding and anti-viral
activity equivalent to that of native griffithsin.
[0154] This example illustrates a method of producing
recombinant griffithsin, which displays physical and functional
properties similar, if not identical, to that of natural
griffithsin.
**Example 4**
[0155] This example describes a method of purifying a
recombinant His-tagged-griffithsin protein.
[0156] Using an immobilized metal affinity chromatography set-up
including Ni-NTA agarose (QIAGEN Inc., Valencia, Calif.), a
His-tagged-griffithsin fusion protein (as described in Example
3) was purified.
[0157] The soluble fraction described in Example 3 was loaded
onto 20 ml gravity columns containing affinity matrix. The
columns were washed extensively with washing buffer (50 mM
NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0) to remove
contaminating proteins. Since His-tagged griffithsin cannot
compete for binding sites on the Ni-NTA resin if the imidazole
concentration is increased to 100-250 mM, the His-tagged
griffithsin protein was eluted by applying elution buffer (50 mM
NaH2PO4, 300 mM NaCl, 250 mM imidazole, pH 8.0) through the
column. Column fractions and wash volumes were monitored by
Western-blot analysis using Penta-His(TM) antibody (QIAGEN Inc.,
Valencia, Calif.) or anti-griffithsin antibody. Fractions
containing the purified His-tagged griffithsin protein were
pooled, dialyzed extensively against distilled water, and
lyophilized.
[0158] Potent cytoprotective and anti-replicative activities of
both natural and His-tagged recombinant griffithsin were
observed using the HIV-1RF strain of HIB in CEM-SS cells. Both
the natural and recombinant griffithsin polypeptides
demonstrated a concentration-dependent inhibition of
virus-induced cell killing. Griffithsin treatment also resulted
in concomitant decreases in supernatant reverse transcriptase
and viral core antigen, p24. Mid-to-high picomolar
concentrations of griffithsin exhibited comparably potent
activity against all of the representative T-tropic laboratory
strains and primary isolates as well as M-tropic primary
isolates. In the antiviral assays, there was little or no
evidence of direct cytotoxicity of griffithsin to the uninfected
control cells at the highest tested concentrations of
griffithsin (78.3 to 783 nM). Griffithsin-pretreated uninfected
CEM-SS cells retained normal susceptibility to HIV infection
after the removal of griffithsin. In contrast, infectivity of
cell-free virus was abolished after pretreatment and removal of
griffithsin. These results indicate that griffithsin is a
virucide. Cocultivation of uninfected and chronically infected
CEM-SS with griffithsin resulted in concentration-dependent
inhibition of cell-cell fusion. Additional binding and fusion
inhibition assay using [beta]-gal indicator cells showed similar
results. Griffithsin inhibited fusion of CD4 [beta]-gal cells
with HL [2/3] cells and also inhibited cell-free HIV-1IIIB
fusion and infection of [beta]-gal cells in a
concentration-dependent manner.
**Example 5**
[0159] This example illustrates the anti-HIV activity of natural
griffithsin polypeptide and His-tagged griffithsin polypeptide.
[0160] Pure proteins were initially evaluated for antiviral
activity using an XTT-tetrazolium anti-HIV assay described
previously (Boyd, in Aids, Etiology, Diagnosis, Treatment And
Prevention (1988), supra; Gustafson et al., J. Med. Chem., 35:
1978-1986 (1992); Weislow (1989), supra; Gulakowski (1991),
supra). A CEM-SS human lymphocytic target cell line was used in
all assays maintained in RPMI 1650 medium (Gibco, Grand Island,
N.Y.), without phenol red, supplemented with 5% fetal bovine
serum, 2 mM L-Glutamine, and 50 mg/ml Gentamicin (complete
medium).
[0161] Exponentially growing cells were pelleted and resuspended
at a concentration of 2.0\*10<5 >cells/ml in complete
medium. The Haitian variant of HIV, HTLV-IIIRF (3.54\*10<6
>SFU/ml), was used throughout. Frozen virus stock solutions
were thawed immediately before use and resuspended in complete
medium to yield 1.2\*10<5>SFU/ml. The appropriate amounts
of the pure proteins for anti-HIV evaluations were dissolved in
H2O-DMSO (3:1), then diluted in complete medium to the desired
initial concentration. All serial drug dilutions, reagent
additions, and plate-to-plate transfers were carried out with an
automated Biomek 1000 Workstation (Beckman Instruments, Palo
Alto, Calif.).
[0162] FIG. 4 summarizes the observed antiviral activities of
native griffithsin from Griffithsia sp. (FIG. 4a) and
recombinant His-tagged-fusion griffithsin (FIG. 4b). Effects of
a range of concentrations of native griffithsin and
HIS-tagged-griffithsin upon CEM-SS cells infected with HIV-1, as
determined after 6 days in culture is illustrated in FIG. 6.
Data points represent the percent of the respective uninfected,
nondrug-treated control values. The two griffithsin polypeptides
demonstrated potent anti-HIV activity with an EC50 in the low
nanomolar range and no significant evidence of direct
cytotoxicity to the host cells at the highest tested
concentrations (up to 1 mM).
**Example 6**
[0163] This example demonstrates that HIV viral envelope gp120
is the principal target for griffithsin.
[0164] To determine the affinity of griffithsin for a series of
protein standards, 100 ng each of gp160, gp120, gp41, sCD4,
bovine IgG, [alpha]-acid glycoprotein, and aprotinin were
subjected to ELISA as previously described (Bokesch et al.,
Biochemistry, 42: 2578-2584 (2003)). Briefly, the protein
standards were bound to a 96-well plate, which was rinsed with
PBST (three times) and blocked with BSA. Between each step of
the protocol, the plate was rinsed with PBST (three times). The
protein standards were incubated with griffithsin (100 ng/well),
followed by incubation with a 1:500 dilution of an
anti-griffithsin rabbit polyclonal antibody preparation.
Griffithsin bound to the protein standards was detected by
adding goat-anti-rabbit antibodies conjugated to alkaline
phosphatase (Roche Molecular Biochemicals, Indianapolis, Ind.).
Upon addition of alkaline phosphatase substrate buffer,
absorbance was measured at 405 nm for each well.
Glycosylation-dependent binding of griffithsin to gp120 was
examined using an ELISA as above, with glycosylated and
nonglycosylated gp120 (HIV-1SF2 gp120) added to the 96-well
plate and incubated with serial dilutions of griffithsin.
[0165] Griffithsin was tested for its ability to bind HIV
envelope glycoproteins. Evidence for direct interaction of
griffithsin with gp120, gp160, and to a lesser degree, gp41 was
obtained from ELISA experiments (FIG. 5a). There was little or
no detectable interaction between griffithsin and cCD4 or other
reference proteins, including bovine IgG, [alpha]-acid
glycoprotein, and aprotinin. An additional ELISA experiment
showed that binding of griffithsin to sgp120 is both
concentration-dependent and glycosylation-dependent (FIG. 5b).
[0166] To undertake preliminary mapping studies to define
griffithsin-binding site on the gp120, we evaluated the effect
of griffithsin on the reactivity of soluble CD4 (sCD4),
cyanovirin-N, and a panel of monoclonal antibodies (mAb) with
soluble gp120 (sgp120) in an ELISA format assay. These studies
demonstrated that griffithsin interfered strongly with
recognition of sgp120 by the mAbs 48d and 2G12. Griffithsin
moderately interfered with sCD4 and mAb IgG1b12 binding to
sgp120. Griffithsin had little or no effect on the recognition
of sgp120 by mAbs that recognize the C1 region (or V3 loop), and
the mAb 17b. However, additional studies demonstrated that
pretreatment of sgp120 with sCD4 and the mAbs IgG 1b12, 48d, and
2G12 did not block subsequent binding of griffithsin to sgp120.
Cyanovirin-N interfered strongly with the recognition of sgp120
by griffithsin. On the other hand, griffithsin pretreatment of
sgp120 did not block subsequent binding of cyanovirin-N to
sgp120.
[0167] Since griffithsin inhibited viral entry, we compared
matched control and griffithsin-treated sgp120 preparations in a
flow cytometric sgp120/CD4-expressing cell binding assay to
determine whether griffithsin inhibits viral attachment or
subsequent fusion events. The CEM-SS cell line expresses CD4, as
demonstrated by the binding of target cells with both anti-Leu3a
and anti-OKT4 monoclonal antibodies. After incubation of CEM-SS
cells with sgp120, the cells were stained by anti-gp120
mAb-FITC. A concomitant decrease in the availability of the
Leu3a epitope (i.e., the gp120-binding site on target cells) was
observed. In other words, the sgp120 bound to the gp120 binding
site on the target cells. As expected, little change in the
staining specific for the OKT4 epitope (i.e., a non-gp120
binding site) was observed. These results are consistent with
sgp120 binding of CD4 on the target cells. Pretreatment of
sgp120 with griffithsin substantially recovered the availability
of the Leu3a epitope, indicating that griffithsin completely
blocked CD4-dependent sgp120 binding. However, overall sgp120
binding showed two peaks in the flow cytometry data when
griffithsin-treated sgp120 was added to the cells. The decreased
signal suggests inhibition of sgp120 binding to CD4 by
griffithsin, which was consistent with the recovery of the
availability of the Leu3a epitope. The increased signal suggests
that the griffithsin/sgp120 complex also non-specifically bound
to target cells.
[0168] This example demonstrates that griffithsin binds to a
region of gp120 that recognizes CD4 on host cells.
**Example 7**
[0169] This example illustrates the broad-range anti-HIV
activity of griffithsin.
[0170] Anti-viral assays used to study the activities of
laboratory strains and primary isolates of virus have been
previously published (Buckheit et al., Antiviral Res., 21:
247-265 (1993)). The low passage HIV-1 pediatric isolate ROJO
was derived as previously described (Buckheit et al., AIDS Res.
Hum. Retroviruses, 10: 1497-1506 (1994)). Peripheral blood
mononuclear cells (PBMC) and macrophages were isolated from
hepatitis and HIV sero-negative donors following Ficoll-Hypaque
centrifugation as described elsewhere (Gartner and Popovic,
Techniques in HIV Research, Aldovini, A. and Walker, B., eds.,
Stockton Press, New York (1994) pp. 59-63). Mean EC50 values
were determined from concentration-response curves from eight
dilutions of griffithsin (triplicate wells/concentration);
assays for HIV-1 RF/CEM-SS employed XTT-tetrazolium; HIV-1 ROJO
were tested in human PBMC cultures by supernatant reverse
transcriptase activity; HIV-1 Ba-L and ADA were tested in human
primary macrophage cultures by p24 ELISA assay. Standard errors
averaged less than 10% of the respective means. The results of
this study are summarized in Table 1 below.
[0000]
TABLE 1
Virus Target Cell Tropism EC50 (nM)
HIV-1 Laboratory
Strain
RF CEM-SS T 0.043
HIV Primary Isolates
ROJO PBMC T 0.63
ADA Macrophage M 0.50
Ba-L Macrophage M 0.098
[0171] The results show that griffithsin is potently active
(sub-nanomolar EC50 values) against a broad range of HIV
isolates including T-tropic viruses (utilizing CCR5 as a
co-receptor) and M-tropic viruses (utilizing CXCR4 as a
co-receptor). This picomolar level of activity is more potent
than that described for most of the current anti-HIV agents
utilized in therapy or in development, including the entry
inhibitors cyanovirin-N and Enfurtide(R). The data also show
that griffithsin is effective at inhibiting infection by both
laboratory-adapted strains and, more importantly, primary
clinical isolates of HIV (e.g., ROJO, ADA, and Ba-L). Finally,
the results indicate that griffithsin is active regardless of
the cell type used in the assay, having potent activity whether
the cells were T-lymphocytes (CEM-SS), PBMCs, or macrophages.
Griffithsin did not show any toxicity against any of the cell
lines even at concentrations 1000-fold higher than the EC50
values.
**Example 8**
[0172] This example describes the production of anti-griffithsin
polyclonal antibodies. A flow diagram illustrating a method of
producing anti-griffithsin antibodies is provided in FIG. 6.
[0173] A New Zealand white rabbit was immunized with 100 [mu]g
of griffithsin in Freund's complete adjuvant. Booster injections
of 50 [mu]g of griffithsin in Freund's incomplete adjuvant were
administered on days 13, 29, 51, 64, 100, and 195. On days 7,
21, 42, 63, 78, and 112, 10 mL of blood was removed from the
rabbit. On day 112 the rabbit was sacrificed and bled out. The
IgG fraction of the immune sera of the rabbit was isolated by
protein-A Sepharose affinity chromatography (Bio-Rad, Hercules,
Calif.) according to the manufacturer's instructions. Reactivity
of the polyclonal antibodies for griffithsin was demonstrated by
immunoblot and ELISA studies with 1:500 to 1:3000 dilution of
the rabbit immunoglobulin fractions.
[0174] For immunoblotting, samples were transferred to PVDF
membranes following SDS-PAGE according to standard procedures.
The membranes were incubated for 1 hour with anti-griffithsin
polyclonal antibodies, washed three times with PBS containing
0.05% Tween 20 (PBST), and then treated with goat anti-rabbit
IgG antibodies conjugated to horseradish peroxidase (Sigma, St.
Louis, Mo.). After three washes with PBST, bound antibodies were
visualized by incubating membranes in a solution of 0.05%
3,3'-diaminobenzidine and 0.003% H2O2.
[0175] The IgG fraction of rabbit polyclonal anti-griffithsin
antibodies were purified after the final boost and animal
sacrifice by using protein-A Sepharose chromatography on the 57
mL of rabbit serum collected. Following purification, 78 mL of
purified anti-griffithsin IgGs were produced. The final
concentration of protein was 335 micrograms/mL for a total yield
of 27.3 mg of anti-griffithsin IgG. To analyze the specificity
of the resulting antibody preparation, Western blot analysis was
performed and resulted in the clear determination of specificity
and avidity for griffithsin by the purified antibodies. A 1:250
dilution of the purified antibodies clearly visualized only the
griffithsin from a mixture of griffithsin and other proteins.
The response to griffithsin by the anti-griffithsin antibodies
was also shown to be concentration-dependent.
**Example 9**
[0176] This example illustrates the anti-influenza virus
activity of griffithsin.
[0177] All examined influenza viruses were passaged in Madin
Darby canine kidney (MDCK) cells to prepare viral stocks. MDCK
cells (from ATCC, Manassas, Va.) were grown in antibiotic-free
minimum essential medium (MEM) with non-essential amino acids
(Gibco, Long Island, N.Y.) containing 5% fetal bovine serum
(FBS, HyClone Laboratories, Logan, Utah) and 0.1% NaHCO3. Test
medium consisted of MEM with 0.18% NaHCO3, 10 units/mL trypsin,
1 [mu]g of ethylenediaminetetraacetate (EDTA) per ml, and 50
[mu]g gentamicin/mL.
[0178] Inhibition of virus-induced cytopathic effect (CPE) as
determined by visual (microscopic) examination of infected cells
and confirmed by increase in neutral red (NR) dye uptake into
infected cells was used as an indicator of griffithsin antiviral
activity. The CPE inhibition method was reported previously by
Smee et al. (Antiviral Res., 5: 251-259 (2001)). Seven
concentrations of griffithsin were screened for antiviral
activity against each virus in 96-well flat-bottomed microplates
of cells. The griffithsin protein was added 5-10 minutes prior
to addition of virus to the cells. The concentration of virus
correspond to approximately 50% infection of cells in culture
(CCID50) per well. The virus challenge dose equals a
multiplicity of infection of approximately 0.001 infectious
particles per cell. The reaction proceeded at 37[deg.] C. for 72
hr. To perform the NR uptake assay for confirmation of antiviral
activity, dye (0.34% concentration in medium) was added to the
plates used to obtain visual scores of CPE. After 2 hours, color
intensity of the dye absorbed by and subsequently eluted from
the cells was determined by the method of Finter et al., J. Gen.
Virol., 5, 419-427 (1969) using a computerized EL-309 microplate
autoreader (Bio-Tek Instruments, Winooski, Vt.). Antiviral
activity was expressed as the 50% effective (virus-inhibitory)
concentration (EC50 value) determined by plotting griffithsin
concentration versus percent inhibition on semi-logarithmic
graph paper. Cytotoxicity of compounds was assessed in parallel
with the antiviral determinations in the same microplates,
except in the absence of virus. From these, 50% cytotoxic
endpoints (IC50 values) were determined. The results of this
study are summarized in Table 2.
[0000]
**TABLE 2**
Influenza Virus Strain EC50 ([mu]g/ml)
Beijing/262/95 (H1N1) 0.07
Texas/36/91 (H1N1) 0.06
Los Angeles/2/87 (H3N2) 0.037
Panama/2007/99 (H3N2) 0.006
Shandong/09/93 (H3N2) 0.018
Sydney/5/97 (H3N2) 0.016
Washington/05/96 (H3N2) 0.016
[0179] Similar to the results with HIV, griffithsin was found to
be potently active against a wide spectrum of influenza A
viruses. These viruses included both H1N1 strains and H3N2
strains of influenza, which is especially significant in light
of the fact that the highly virulent Fijian strain of influenza
A that afflicted the United States in 2003/2004 was also a H3N2
strain. Griffithsin was not found to be toxic to the MDCK cell
line utilized for these experiments, even when the cells were
exposed to a high dose of griffithsin (10 micrograms/mL).
**Example 10**
[0180] This example describes a method of producing recombinant
griffithsin.
[0181] Recombinant expression of His-tagged griffithsin in E.
coli was optimized using a fermenter in combination with an
auto-induction media. A seed culture was grown in LB media
containing 30 [mu]g/ml kanamycin in a shaker flask at 37[deg.]
C. and 150 rpm for 17 hours. In addition, a fermenter containing
an auto-induction media was inoculated with the seed culture.
The ratio of auto-induction media to seed culture was
approximately 50:1. The culture was grown at 37[deg.] C. for 24
hours. The final culture density was approximately 8.6 OD600
units. The final culture was harvested by centrifugation, and
the soluble fraction was obtained as described above.
[0182] Crude soluble fractions contained His-tagged-griffithsin
fusion protein, which was detected by Western-blotting with
anti-griffithsin polyclonal antibodies. The ratio of
soluble:insoluble protein at approximately 15 kDa was 50:50. The
ratio indicates that more griffithsin protein was produced in
soluble fraction in this fermentation procedure compared with
protein expression achieved using a shaker flask procedure. In
addition, the fermentation procedure provided approximately
30-fold higher quantities of griffithsin protein than the shaker
flask procedure. Approximately 50 mg of purified recombinant
griffithsin was isolated from 1 L of the fermentation. The
purified protein existed as a homodimer and demonstrated gp120
binding and anti-viral activity equivalent to that of native
griffithsin.
[0183] The results of this example confirm a method of producing
recombinant, anti-viral griffithsin protein.
[0184] The following references, to the extent that they provide
exemplary procedural or other details supplementary to those set
forth herein, are specifically incorporated herein by reference:
Birren et al., Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, N.Y. (1997),
Birren et al., Genome Analysis: A Laboratory Manual Series,
Volume 2, Detecting Genes, Cold Spring Harbor Laboratory Press,
Cold Spring Harbor, N.Y. (1998),
Birren et al., Genome Analysis: A Laboratory Manual Series,
Volume 3, Cloning Systems, Cold Spring Harbor Laboratory Press,
Cold Spring Harbor, N.Y. (1999),
Birren et al., Genome Analysis: A Laboratory Manual Series,
Volume 4, Mapping Genomes, Cold Spring Harbor Laboratory Press,
Cold Spring Harbor, N.Y. (1999),
Harlow et al., Antibodies: A Laboratory Manual, Cold Spring
Harbor Laboratory Press, Cold Spring Harbor, N.Y. (1988),
Harlow et al., Using Antibodies: A Laboratory Manual, Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. (1999),
and
Sambrook et al., Molecular Cloning: A Laboratory Manual, 2nd
edition, Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, N.Y. (1989).
---
**US8088729**
**Anti-viral griffithsin compounds, compositions, and
methods of use**
A method of inhibiting a viral infection of
a host comprising administering to the host an anti-viral
griffithsin polypeptide comprising SEQ ID NO: 3 or a
fragment thereof comprising at least eight contiguous amino
acids, a nucleic acid encoding the anti-viral polypeptide,
or an antibody to the anti-viral polypeptide. A method of
inhibiting a virus in a sample comprising contacting the
sample with an anti-viral griffithsin polypeptide or
antibody thereto also is provided.
**TECHNICAL FIELD OF THE INVENTION**
The invention relates to an anti-viral Griffithsin
polypeptide related conjugates, compositions, nucleic acids,
vectors, host cells, antibodies, and methods for their
production and use.
**BACKGROUND OF THE INVENTION**
Although the field of viral therapeutics has advanced in
response to the need for treatments and prophylactics
effective against diverse classes of viruses, the threat of
viruses remain among several human populations across the
world.
Retroviruses, such as HIV, continue to pose a threat to
humans. There are many ways in which an agent can exhibit
anti-retroviral activity (e.g., see DeClercq, Adv. Virus
Res., 42: 1-55 (1993); DeClercq, J. Acquir. Immun. Def.
Synd., 4: 207-218 (1991); and Mitsuya et al., Science, 249:
1533-1544 (1990). Nucleoside derivatives, such as AZT, which
inhibit the viral reverse transcriptase, were among the
first clinically active agents available commercially for
anti-HIV therapy. Although very useful in some patients, the
utility of AZT and related compounds is limited by toxicity
and insufficient therapeutic indices for fully adequate
therapy. Also, given the subsequent revelations about the
true dynamics of HIV infection (Coffin, Science, 267:
483-489 (1995); and Cohen, Science, 267: 179 (1995)), it has
become increasingly apparent that agents acting as early as
possible in the viral replicative cycle are needed to
inhibit infection of newly produced, uninfected immune cells
generated in the body in response to the virus-induced
killing of infected cells. Also, it is essential to
neutralize or inhibit new infectious virus produced by
infected cells.
Effective means for preventing HIV infection also are needed
as a global priority. Heterosexual transmission accounts for
the majority of new cases of HIV infection each year.
Current reports from the World Health Organization estimate
that a total of more than 40 million people are now infected
with HIV. HIV prevention research has to date focused
predominantly on vaccine development. However, no effective
preventative or therapeutic vaccine has been identified thus
far. New approaches to vaccine development, as well as
entirely different strategies and agents for preventing
person-to-person transmission of HIV infection, are needed.
One approach showing great promise is the development and
use of topical microbicides. In this approach, a suitable
antiviral agent is applied directly at the potential site of
virus exposure, e.g., the genital mucosa in the case of HIV.
A suitable antiviral agent is one which inactivates or
inhibits infectivity of a virus upon contact of the
antiviral agent with the virus. Suitable animal models are
available for demonstrating in vivo efficacy of such
approaches for preventing transmission of immunodeficiency
viruses, such as HIV. For instance, the HIV-inactivating
protein, cyanovirin-N, has been shown to inhibit the sexual
transmission of a chimeric simian/human immunodeficiency
virus (SHIV) infection in a primate model employing macaques
exposed to the virus vaginally or rectally (C-C Tsai et al.,
AIDS Res. Hum. Retroviruses, 19, 535-541 (2003) and C-C Tsai
et al., AIDS Res. Hum. Retroviruses, 20, 11-18 (2004)).
Infection of people by influenza viruses is also a major
cause of pandemic illness, morbidity and mortality
worldwide. The adverse economic consequences, as well as
human suffering, are enormous. Available treatments for
established infection by this virus are either minimally
effective or ineffective; these treatments employ
amantatadine, rimantadine and neuraminidase inhibitors. Of
these drugs, only the neuraminidase inhibitors are
substantially active against multiple strains of influenza
virus that commonly infect humans, yet these drugs still
have limited utility or efficacy against pandemic disease.
Currently, the only effective preventative treatment against
influenza viral infection is vaccination. However, this,
like the drug treatments, is severely limited by the
propensity of influenza viruses to mutate rapidly by genetic
exchange, resulting in the emergence of highly resistant
viral strains that rapidly infect and spread throughout
susceptible populations. In fact, a vaccination strategy is
only effective from year-to-year if the potential pandemic
strains can be identified or predicted, and corresponding
vaccines prepared and administered early enough that the
year's potential pandemic can be aborted or attenuated.
Thus, new preventative and therapeutic interventions and
agents are urgently needed to combat influenza viruses.
New agents with broad anti-influenza virus activity against
diverse strains, clinical isolates and subtypes of influenza
virus would be highly useful, since such agents would most
likely remain active against the mutating virus. The two
major types of influenza virus that infect humans are
influenza A and B, both of which cause severe acute illness
that may include both respiratory and gastrointestinal
distress, as well as other serious pathological sequellae.
An agent that has anti-influenza virus activity against
diverse strains and isolates of both influenza A and B,
including recent clinical isolates thereof, would be
particularly advantageous for use in prevention or treatment
of hosts susceptible to influenza virus infection.
The predominant mode of transmission of influenza viral
infection is respiratory, i.e., transmission via inhalation
of virus-laden aerosolized particles generated through
coughing, sneezing, breathing, etc., of an
influenza-infected individual. Transmission of infectious
influenza virions may also occur through contact (e.g.,
through inadvertent hand-to-mouth contact, kissing,
touching, etc.) with saliva or other bodily secretions of an
infected individual. Thus, the primary first points of
contact of infectious influenza virions within a susceptible
individual are the mucosal surfaces within the oropharyngeal
mucosa, and the mucosal surfaces within the upper and lower
respiratory tracts. Not only do these sites comprise first
points of virus contact for initial infection of an
individual, they are also the primary sites for production
and exit (e.g., by coughing, sneezing, salivary
transmission, etc.) of bodily fluids containing infectious
influenza viral particles. Therefore, availability of a
highly potent anti-influenza virus agent, having
broad-spectrum activity against diverse strains and isolates
of influenza viruses A and B, which could be applied or
delivered topically to the aforementioned mucosal sites of
contact and infection and transmission of infectious
influenza viruses, would be highly advantageous for
therapeutic and preventative inhibition of influenza viral
infection, either in susceptible uninfected or infected
hosts.
Highly pathogenic avian H5N1 influenza A viruses have been
of widespread concern in recent years. The H5N1 virus can be
highly lethal to birds and humans raising concerns of a
possible pandemic (Hatta and Kawaoka, Uirusu. 55(1):55-61
(2005)). The well-established pathogenicity of these avian
influenza viruses makes evident the need in the art for the
development of effective anti-H5N1 drugs and vaccines.
Infection with hepatitis C virus (HCV) also represents an
important public health problem. HCV is a leading cause of
chronic hepatitis, cirrhosis, and hepatocellular carcinoma
(Verslype et al., Acta Gastroenterol Belg. 68(3):314-318
(2005)). Patients with HCV are mainly treated today with
interferon, alone or in combination with ribavirin. However,
such treatments eliminate the virus from only about one half
of the patients (Watashi and Shimotohno, Uirusu.
55(1):105-110 (2005)). Therefore, a more effective approach
to the treatment of HCV infection is needed.
In the latter part of 2002, a new disease, severe acute
respiratory syndrome (SARS), emerged in China, and an animal
coronavirus that had crossed the species barrier through
close contact of humans with infected animals was later
identified as the etiological agent. The coronavirus rapidly
adapted to the new host and not only became readily
transmissible between humans but also more pathogenic. Air
travel spread the virus rapidly around the world and
ultimately the virus infected 8096 people and caused 774
deaths in 26 countries on 5 continents. Aggressive
quarantine measures successfully terminated SARS (Stadler
and Rappuoli, Curr Mol Med. 5(7):677-697 (2005)). However, a
resurgence of SARS is still a threat, because the causative
agent remaining in animal reservoirs is not fully
understood, and sporadic cases continue to be reported (Lu
et al., Acta Pharmacol Sin. 26(12):1479-1484 (2005)).
Therefore, there is a need in the art to develop antiviral
drugs and vaccines specific for the SARS virus.
The Zaire ebola virus has caused large outbreaks of severe
and usually fatal hemorrhagic disease in humans for which
there is no effective treatment or cure (Towner et al.,
Virology 332(1):20-27 (2005)). Thus, there is a need in the
art for effective methods of treating or preventing ebola
viral infections in humans.
In this regard, new classes of anti-viral agents, to be used
alone or in combination existing anti-viral agents, are
needed for effective anti-viral therapy. New agents are also
important for the prophylactic inhibition of viral
infection. In both areas of need, the ideal new agent(s)
would act as early as possible in the viral life cycle; be
as virus-specific as possible (i.e., attack a molecular
target specific to the virus but not the host); render the
intact virus noninfectious; prevent the death or dysfunction
of virus-infected cells; prevent further production of virus
from infected cells; prevent spread of virus infection to
uninfected cells; be highly potent and active against the
broadest possible range of strains and isolates of a given
virus; be resistant to degradation under physiological and
rigorous environmental conditions; and be readily and
inexpensively produced.
In view of the foregoing, there is a need in the art for new
methods and compositions for inhibiting viral infection. The
invention provides such methods. These and other advantages
of the invention, as well as additional inventive features,
will become apparent from the description provided herein.
**BRIEF SUMMARY OF THE INVENTION**
The invention provides, among other things, an isolated and
purified nucleic acid molecule that encodes a polypeptide
comprising at least eight contiguous amino acids of SEQ ID
NO: 3, wherein the at least eight contiguous amino acids
have anti-viral activity, optionally as part of an encoded
fusion protein. In this regard, the invention also provides
an isolated and purified nucleic acid molecule that encodes
a polypeptide comprising at least eight contiguous amino
acids of SEQ ID NO: 3, wherein the at least eight contiguous
amino acids comprise amino acids 1-121 of SEQ ID NO: 3 which
have been rendered glycosylation-resistant and wherein the
at least eight contiguous amino acids have antiviral
activity, optionally as part of an encoded fusion protein.
Further provided are vectors comprising an aforementioned
isolated and purified nucleic acid molecule and a host cell
or organism comprising such a vector.
Accordingly, the invention also provides a method of
producing an anti-viral polypeptide, which method comprises
expressing the nucleic acid molecule, optionally in the form
of a vector, in a host cell or organism. Thus, an anti-viral
polypeptide comprising at least eight contiguous amino acids
of SEQ ID NO: 3, wherein the at least eight contiguous amino
acids have anti-viral activity, and an antiviral polypeptide
comprising at least eight contiguous amino acids of SEQ ID
NO: 3, wherein the at least eight contiguous amino acids
comprise amino acids 1-121 of SEQ ID NO: 3, which have been
rendered glycosylation-resistant and wherein the at least
eight contiguous amino acids have antiviral activity, are
also provided, as are conjugates comprising an
aforementioned anti-viral polypeptide and at least one
effector component. Compositions comprising an effective
amount of an aforementioned anti-viral polypeptide or
anti-viral polypeptide conjugate are also provided.
The invention further provides a method of inhibiting
prophylactically or therapeutically a viral infection of a
host, such as a retroviral infection of a host (e.g., human
immunodeficiency virus (HIV), e.g., HIV-1 or HIV-2) or,
especially, a viral infection by an influenza virus (e.g.,
an H5N1 virus), Severe Acute Respiratory Syndrome (SARS)
virus, Hepatitis C virus, or Ebola virus. The method
comprises administering to the host an effective amount of
an anti-viral polypeptide or anti-viral polypeptide
conjugate as described herein (e.g., comprising SEQ ID NO:3
or antiviral fragment thereof comprising at least eight
contiguous amino acids of SEQ ID NO: 3), whereupon the viral
infection is inhibited.
Still further provided is a method of inhibiting
prophylactically or therapeutically a viral infection of a
host, e.g., an animal, comprising transforming host cells in
vivo with a nucleic acid molecule encoding an
above-described polypeptide. Even still further provided is
a method of inhibiting prophylactically or therapeutically a
viral infection of a host, e.g., an animal, comprising
transforming host cells with a nucleic acid molecule
encoding an above-described polypeptide and placing the
transformed host cells into or onto the host.
The present invention also provides a method of removing
virus from a sample. The method comprises contacting the
sample with a composition comprising an anti-viral
polypeptide or conjugate or fusion protein thereof, wherein
the anti-viral polypeptide comprises at least eight
contiguous amino acids of SEQ ID NO: 3, which at least eight
contiguous amino acids of SEQ ID NO: 3 bind to the virus.
An antibody that binds Griffithsin is provided as is a
composition comprising the same. Similarly, an
anti-Griffithsin antibody is provided as is a composition
comprising the same. A method of administering an
anti-Griffithsin antibody or a composition comprising the
same to a mammal so as to inhibit infection of the mammal
with a virus is also provided.
**BRIEF DESCRIPTION OF THE DRAWINGS****FIG. 1 is a flow diagram illustrating an anti-HIV
bioassay-guided method of isolating, purifying, and
elucidating the amino acid sequence of Griffithsin.****FIG. 2 is a flow diagram illustrating a method of
synthesizing a recombinant Griffithsin gene.****FIG. 3 is a flow diagram illustrating a method of
expressing a synthetic Griffithsin gene encoding a
His-tagged Griffithsin polypeptide protein and
purification of the recombinant His-tagged Griffithsin.****FIG. 4a is a line graph illustrating the anti-HIV
activity of native Griffithsin, in terms of concentration
of Griffithsin (nM) (X-axis) versus % control (Y-axis).
FIG. 4b is a line graph illustrating the anti-HIV activity
of recombinant, His-tagged Griffithsin in terms of
concentration of Griffithsin (nM) (X-axis) versus %
control (Y-axis).****FIG. 5a is a bar graph comparing test proteins bound
by Griffithsin (Y-axis) and absorbance of the
Griffithsin-test protein complex at 405 nm (X-axis). FIG.
5b illustrates the concentration-dependent binding of
Griffithsin to gp120 by comparing Griffithsin (GRFT)
concentration (pmol) and absorbance of Griffithsin-gp120
complexes at 405 nm.****FIG. 6 is a flow diagram illustrating a method of
producing anti-Griffithsin antibodies.****FIG. 7 is a graph of the anti-viral effect (% virus
control; -) or the cytotoxic effect (% cell viability; -)
of Griffithsin at different concentrations.**
**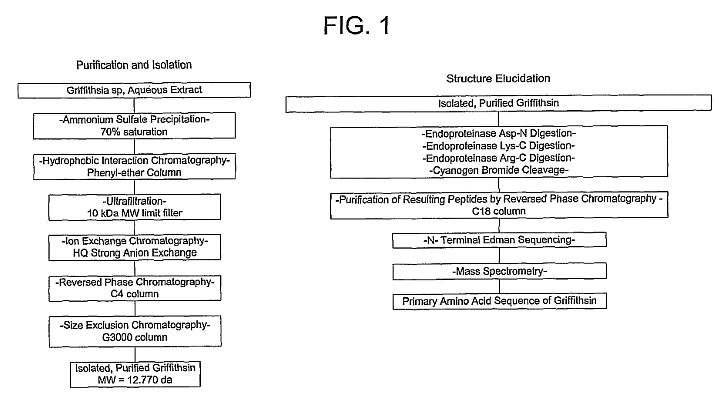 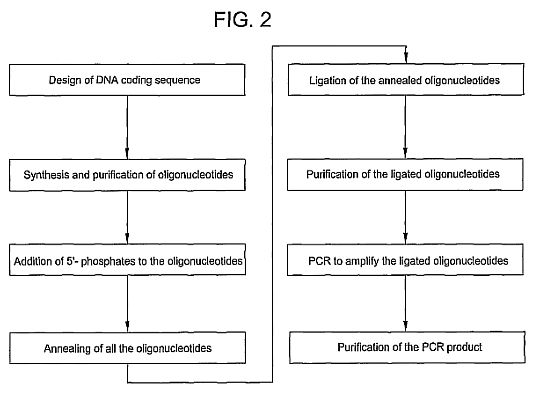 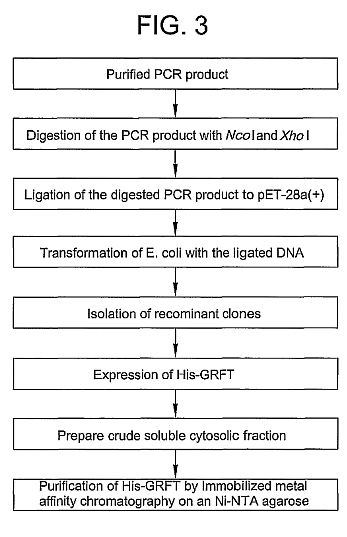 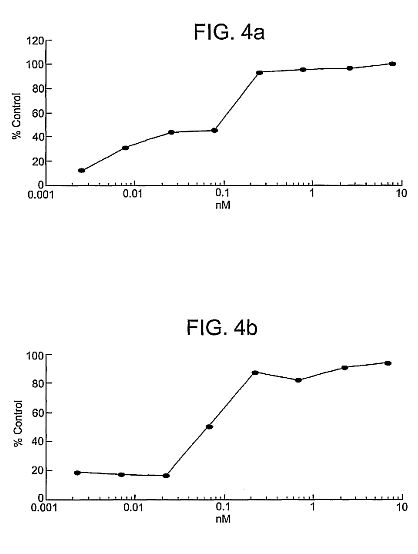 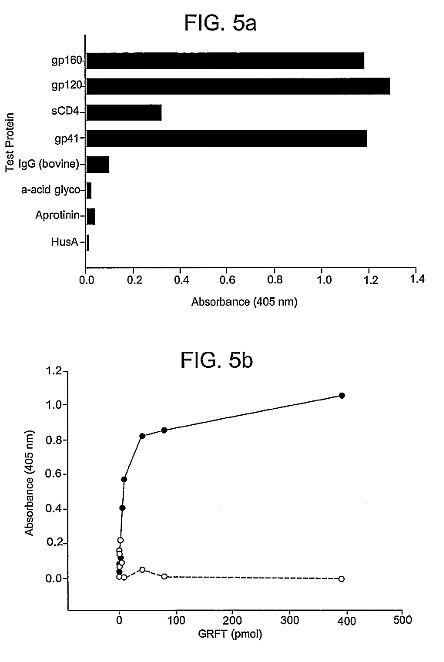 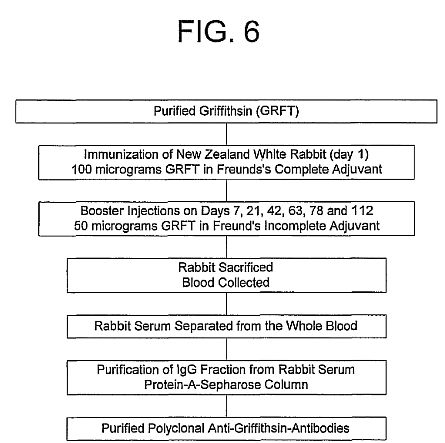** **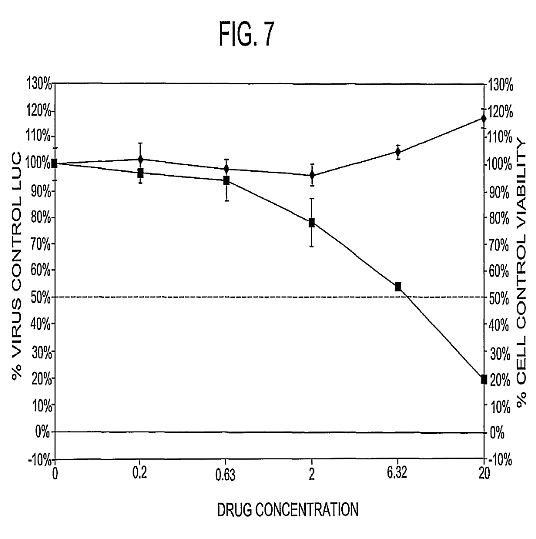****DETAILED DESCRIPTION OF THE PRESENT INVENTION**
The principal overall objective of the invention is to
provide an anti-viral polypeptide and derivatives thereof,
and broad uses thereof (e.g., medical and research uses),
including prophylactic and/or therapeutic applications
against viruses. An initial observation, which led to the
invention, was anti-viral activity of certain extracts from
a marine organism, namely Rhodophyte (Griffithsia sp.),
originally collected in the territorial waters of New
Zealand. Low picomolar concentrations of a protein isolated
from the extracts, referred to herein as Griffithsin,
irreversibly inactivated human clinical isolates of HIV. Its
HIV molecular target is high mannose-comprised
oligosaccharide constituents of Env glycoproteins. Upon
binding, Griffithsin inhibits viral binding, fusion, and
entry. Griffithsin also targets other viruses, such as other
retroviruses, e.g., EV, SIV and HTLV, and non-retroviruses,
such as measles and, especially, influenza (e.g., H5N1
virus), Ebola, Hepatitis C, and SARS virus.
Accordingly, the invention provides an isolated and purified
anti-viral polypeptide of SEQ ID NO: 3 from Griffithsia sp.
and functional homologs thereof, referred to collectively as
"Griffithsin." Herein the term "Griffithsin" is used
generically to refer to a natural Griffithsin or any
related, functionally equivalent (i.e., anti-viral)
polypeptide or derivative thereof. By definition, in this
context, a related, functionally equivalent polypeptide or
derivative thereof (a) contains a sequence of at least eight
contiguous amino acids directly identical to a sub-sequence
of eight contiguous amino acids contained within a natural
Griffithsin, and (b) can specifically bind to a virus, in
particular an influenza virus (e.g., H5N1), Hepatitis C, or
SARS, Ebola, a retrovirus, more specifically a primate
immunodeficiency virus, more specifically HIV-1, HIV-2 or
SIV, or to an infected host cell expressing one or more
viral antigen(s), more specifically an envelope
glycoprotein, such as gp120, of the respective virus. In
addition, such a functionally equivalent polypeptide or
derivative thereof can comprise the amino acid sequence of a
natural Griffithsin (see SEQ ID NO: 3), in which 1-20,
preferably 1-10, more preferably 1, 2, 3, 4, or 5, and most
preferably 1 or 2, amino acids have been removed from one or
both ends, preferably from only one end, e.g., removed from
the amino-terminal end, of natural Griffithsin.
Alternatively, a functionally equivalent polypeptide or
derivative thereof can comprise the amino acid sequence of a
native Griffithsin (see SEQ ID NO: 3), in which 1-20,
preferably 1-10, more preferably 1, 2, 3, 4, or 5, and most
preferably 1 or 2, amino acids have been added to one or
both ends, preferably from only one end, e.g., the
amino-terminal end, of the native Griffithsin.
The invention further provides an isolated and purified
polypeptide encoded by a nucleic acid molecule comprising a
sequence of SEQ ID NO: 1 or a nucleic acid molecule encoding
an amino acid sequence of SEQ ID NO: 2 or SEQ ID NO: 3. Upon
examination of the antiviral Griffithsin polypeptide, the
amino acid at position 31 of SEQ ID NO: 3 (represented as
Xaa) was found not to be a familiar amino acid residue.
Placement of an alanine at position 31, such as achieved in
the recombinant Griffithsin polypeptide described herein
(SEQ ID NO: 2), results in a polypeptide exhibiting
equivalent activity as the natural Griffithsin polypeptide.
If desired, the amino acid at position 31 can be substituted
with any other amino acid to facilitate protein production.
Ideally, the substitution at position 31 of SEQ ID NO: 3
does not diminish the anti-viral activity of the protein
(e.g., does not diminish the anti-viral activity more than
50%, more than 30% or more than 10%) as compared to the
anti-viral activity of the native protein. Preferably, the
aforementioned nucleic acid molecules encode at least eight
(e.g., at least 10, at least 20, at least 30, at least 50,
at least 70, at least 80, at least 90, or at least 100)
contiguous amino acids of the amino acid sequence of SEQ ID
NO: 3, which desirably have anti-viral activity. If the at
least eight contiguous amino acids of SEQ ID NO: 3 comprise
amino acids 1-121, desirably amino acid residue 45, 60, 71,
and/or 104 has been rendered glycosylation resistant, while
maintaining antiviral activity of the polypeptide.
The term "isolated" as used herein means having been removed
from its natural environment. The term "purified" as used
herein means having been increased in purity, wherein
"purity" is a relative term and not to be construed as
absolute purity. By "antiviral" is meant that the
polypeptide or fragment thereof can inhibit a virus (e.g.,
inhibit entry of a virus into a host cell, limit the spread
of viral infection by inhibiting cell to cell fusion, and
the like), in particular an influenza virus, such as
influenza virus of a strain A or strain B, or an H5N1
influenza virus, a retrovirus, specifically a primate
immunodeficiency virus (e.g., an HIV virus such as HIV-1,
HIV-2 or SW), a SARS coronavirus, Ebola, or a Hepatitis C
virus.
Preferably, the polypeptide or derivative thereof comprises
an amino acid sequence that is substantially homologous to
that of an anti-viral protein from Griffithsia sp. By
"substantially homologous" is meant sufficient homology to
render the polypeptide or derivative thereof anti-viral,
with anti-viral activity characteristic of an anti-viral
protein isolated from Griffithsia sp. At least about 50%
homology (e.g., at least about 60% homology, at least about
65% homology, or at least about 70% homology), preferably at
least about 75% homology (e.g., at least about 80% homology
or at least about 85% homology), and most preferably at
least about 90% homology (e.g., at least about 95% homology)
should exist.
Alterations of the natural amino acid sequence to produce
variant polypeptides can be done by a variety of means known
to those skilled in the art. For instance, amino acid
substitutions can be conveniently introduced into the
polypeptides at the time of synthesis. Alternatively,
site-specific mutations can be introduced by ligating into
an expression vector a synthesized oligonucleotide
comprising the modified site. Alternately,
oligonucleotide-directed, site-specific mutagenesis
procedures can be used, such as disclosed in Walder et al.,
Gene, 42: 133 (1986); Bauer et al., Gene, 37: 73 (1985);
Craik, Biotechniques, 12-19 (January 1995); and U.S. Pat.
Nos. 4,518,584 and 4,737,462.
It is within the skill of the ordinary artisan to select
synthetic and naturally-occurring amino acids that effect
conservative or neutral substitutions for any particular
naturally-occurring amino acids. The ordinarily skilled
artisan desirably will consider the context in which any
particular amino acid substitution is made, in addition to
considering the hydrophobicity or polarity of the
side-chain, the general size of the side chain and the pK
value of side-chains with acidic or basic character under
physiological conditions. For example, lysine, arginine, and
histidine are often suitably substituted for each other, and
more often arginine and histidine. As is known in the art,
this is because all three amino acids have basic side
chains, whereas the pK value for the side-chains of lysine
and arginine are much closer to each other (about 10 and 12)
than to histidine (about 6). Similarly, glycine, alanine,
valine, leucine, and isoleucine are often suitably
substituted for each other, with the proviso that glycine is
frequently not suitably substituted for the other members of
the group. This is because each of these amino acids are
relatively hydrophobic when incorporated into a polypeptide,
but glycine's lack of an [alpha]-carbon allows the phi and
psi angles of rotation (around the [alpha]-carbon) so much
conformational freedom that glycinyl residues can trigger
changes in conformation or secondary structure that do not
often occur when the other amino acids are substituted for
each other. Other groups of amino acids frequently suitably
substituted for each other include, but are not limited to,
the group consisting of glutamic and aspartic acids; the
group consisting of phenylalanine, tyrosine and tryptophan;
and the group consisting of serine, threonine and,
optionally, tyrosine. Additionally, the ordinarily skilled
artisan can readily group synthetic amino acids with
naturally-occurring amino acids.
The ordinarily skilled artisan can generate Griffithsin
mutants or variants by, for example, substituting or
mutating amino acids which are not critical for the
anti-viral function of the polypeptide. Ideally, mutations
that do not modify the electronic or structural environment
of the peptide are generated to retain optimal antiviral
activity. For example, natural Griffithsin forms dimers,
which can be advantageous in some embodiments. Therefore,
alterations which do not disrupt dimer formation can be
preferred. Amino acid residues which are not responsible for
folding or stability of the three-dimensional conformation
of the Griffithsin polypeptide are candidate residues for
mutation. Alternatively or in addition, amino acids which
are not involved in glycoprotein binding can be mutated or
replaced. It is understood that surface hydrophobicity plays
a key role in protein-protein interactions and surface
electrophilicity is important to protein-sugar interactions,
such as the interaction between Griffithsin and viral
proteins. Hydrophobic surface clusters and electrophilic
surface clusters on the Griffithsin peptide or homologs
which suggest regions critical for interaction with the
viral envelope can be mapped using routine methods such as
those disclosed in Bewley et al., Nature Structural Biology,
5(7): 571-578 (1998). Amino acid residues not found either
in electrophilic or hydrophobic surface clusters are likely
not critical for hydrophobicity or electrophilicity of these
clusters and, thus, are appropriate targets for mutation to
create Griffithsin fragments (e.g., anti-viral polypeptides
comprising at least about eight contiguous amino acids of
SEQ ID NO: 2 or SEQ ID NO: 3), variants, mutants, or
homologs (e.g., Griffithsin variants having 80%, 85%, or 90%
homology to SEQ ID NO: 2 or SEQ ID NO: 3) which retain
antiviral activity. If desired, amino acid residues which
are responsible for binding to high-mannose
oligosaccharide-containing glycoproteins on the viral
surface can be mutated to increase the specificity or
affinity of glycoprotein binding.
If desired, the proteins and peptides of the invention
(including antiviral fragments, variant polypeptides, fusion
proteins, and conjugates) can be modified, for instance, by
glycosylation, amidation, carboxylation, or phosphorylation,
or by the creation of acid addition salts, amides, esters,
in particular C-terminal esters, and N-acyl derivatives of
the proteins of the invention. The polypeptides also can be
modified to create protein derivatives by forming covalent
or noncovalent complexes with other moieties in accordance
with methods known in the art. Covalently-bound complexes
can be prepared by linking the chemical moieties to
functional groups on the side chains of amino acids
comprising the proteins, or at the N- or C-terminus.
Desirably, such modifications and conjugations do not
adversely affect the activity of the polypeptides (and
variants thereof). While such modifications and conjugations
can have greater or lesser activity, the activity desirably
is not negated and is characteristic of the unaltered
polypeptide.
The polypeptides (and fragments, homologs, variants, and
fusion proteins) can be prepared by any of a number of
conventional techniques. The polypeptide can be isolated or
purified from a naturally occurring source or from a
recombinant source. For instance, in the case of recombinant
proteins, a DNA fragment encoding a desired polypeptide can
be subcloned into an appropriate vector using well-known
molecular genetic techniques (see, e.g., Maniatis et al.,
Molecular Cloning: A Laboratory Manual, 2nd ed. (Cold Spring
Harbor Laboratory (1989)) and other references cited herein
under "EXAMPLES"). The fragment can be transcribed and the
polypeptide subsequently translated in vitro. Commercially
available kits also can be employed (e.g., such as
manufactured by Clontech, Palo Alto, Calif.; Amersham Life
Sciences, Inc., Arlington Heights, Ill.; InVitrogen, San
Diego, Calif.; and the like). The polymerase chain reaction
optionally can be employed in the manipulation of nucleic
acids.
Such polypeptides also can be synthesized using an automated
peptide synthesizer in accordance with methods known in the
art. Alternately, the polypeptide (and fragments, homologs,
variants, and fusion proteins) can be synthesized using
standard peptide synthesizing techniques well-known to those
of skill in the art (e.g., as summarized in Bodanszky,
Principles of Peptide Synthesis, (Springer-Verlag,
Heidelberg: 1984)). In particular, the polypeptide can be
synthesized using the procedure of solid-phase synthesis
(see, e.g., Merrifield, J. Am. Chem. Soc., 85: 2149-54
(1963); Barany et al., Int. J. Peptide Protein Res., 30:
705-739 (1987); and U.S. Pat. No. 5,424,398). If desired,
this can be done using an automated peptide synthesizer.
Removal of the t-butyloxycarbonyl (t-BOC) or
9-fluorenylmethyloxycarbonyl (Fmoc) amino acid blocking
groups and separation of the polypeptide from the resin can
be accomplished by, for example, acid treatment at reduced
temperature. The protein-containing mixture then can be
extracted, for instance, with diethyl ether, to remove
non-peptidic organic compounds, and the synthesized
polypeptide can be extracted from the resin powder (e.g.,
with about 25% w/v acetic acid). Following the synthesis of
the polypeptide, further purification (e.g., using HPLC)
optionally can be preformed in order to eliminate any
incomplete proteins, polypeptides, peptides or free amino
acids. Amino acid and/or HPLC analysis can be performed on
the synthesized polypeptide to validate its identity. For
other applications according to the invention, it may be
preferable to produce the polypeptide as part of a larger
fusion protein, either by chemical conjugation or through
genetic means, such as are known to those skilled in the
art. In this regard, the invention also provides a fusion
protein comprising the isolated or purified antiviral
polypeptide (or fragment thereof) or variant thereof and one
or more other protein(s) having any desired properties or
effector functions, such as cytotoxic or immunological
properties, or other desired properties, such as to
facilitate isolation, purification, analysis, or stability
of the fusion protein.
A Griffithsin conjugate comprising a Griffithsin coupled to
at least one effector component, which can be the same or
different, is also provided. The effector component can be
polyethylene glycol, dextran, albumin, an immunological
reagent, a toxin, an antiviral agent, or a solid support
matrix. "Immunological reagent" will be used to refer to an
antibody, an antibody fragment (e.g., an F(ab')2, an Fab',
an Fab, an Fv, an sFv, a dsFv, or an Fc antibody fragment),
an immunoglobulin, and an immunological recognition element.
An immunological recognition element is an element, such as
a peptide, e.g., the FLAG sequence of a recombinant
Griffithsin-FLAG fusion protein, which facilitates, through
immunological recognition, isolation and/or purification
and/or analysis of the protein or peptide to which it is
attached. An immunological reagent also can be an
immunogenic peptide, which can be fused to Griffithsin for
enhancing an immune response. In this respect, the invention
provides an anti-viral conjugate comprising a Griffithsin
polypeptide or fragment thereof bound to a virus or viral
envelope glycoprotein. A Griffithsin fusion protein is a
type of Griffithsin conjugate, wherein a Griffithsin is
coupled to one or more other protein(s) having any desired
properties or effector functions, such as cytotoxic or
immunological properties, or other desired properties, such
as to facilitate isolation, purification or analysis of the
fusion protein or increase the stability or in vivo
half-life of the fusion protein. Griffithsin also can be
attached to a chemical moiety which allows recognition,
isolation, purification, and/or analysis of the protein or
peptide. An example of such a chemical moiety is a His tag
of a recombinant Griffithsin-His fusion protein.
A "toxin" can be, for example, Pseudomonas exotoxin. An
"antiviral agent" can be AZT, ddI, ddC, 3TC gancyclovir,
fluorinated dideoxynucleosides, nevirapine, R82913, Ro
31-8959, BI-RJ-70, acyclovir, [alpha]-interferon,
recombinant sCD4, michellamines, calanolides, nonoxynol-9,
gossypol and derivatives thereof, gramicidin, amantatadine,
rimantadine, and neuraminidase inhibitors, and cyanovirin-N
or a functional homolog or derivative thereof (see, for
example, U.S. Pat. No. 5,843,882). A "solid support matrix"
can be a magnetic bead, a flow-through matrix, a sponge, a
stent, a culture plate, or a matrix comprising a
contraceptive device, such as a condom, diaphragm, cervical
cap, vaginal ring or contraceptive sponge. In an alternative
embodiment, a solid support matrix can be an implant for
surgical implantation in a host and, if appropriate, later
removal.
In view of the foregoing, the invention further provides a
composition comprising (i) the isolated or purified
antiviral polypeptide (or fragment thereof), a variant
thereof, a fusion protein of the antiviral polypeptide (or
fragment thereof) or variant thereof, and a conjugate of the
antiviral polypeptide (or fragment thereof) or variant
thereof, and/or (ii) a carrier, excipient or adjuvant
therefor. Preferably, component (i) of the composition is
present in an antiviral effective amount and the carrier is
pharmaceutically acceptable. By "antiviral effective amount"
is meant an amount sufficient to inhibit the infectivity of
the virus.
The carrier can be any of those conventionally used and is
limited only by chemico-physical considerations, such as
solubility and lack of reactivity with the active agent of
the invention, and by the route of administration. It is
preferred that the pharmaceutically acceptable carrier be
one which is chemically inert to the active agent and one
which has no detrimental side effects or toxicity under the
conditions of use. The pharmaceutically acceptable carriers
described herein, for example, vehicles, adjuvants,
excipients, and diluents, are well-known to those ordinarily
skilled in the art and are readily available to the public.
Typically, the composition, such as a pharmaceutical
composition, can comprise a physiological saline solution;
dextrose or other saccharide solution; or ethylene,
propylene, polyethylene, or other glycol. The pharmaceutical
composition preferably does not comprise mannose or
N-acetyl-glucosamine, as these molecules may interfere with
the functioning of the antiviral agent.
The invention also provides a method of obtaining a
Griffithsin from Griffithsia sp. Such a method comprises (a)
identifying an extract of Griffithsia sp. containing
anti-viral activity, (b) optionally removing high molecular
weight biopolymers from the extract, (c) anti-viral
bioassay-guided fractionating the extract to obtain a crude
extract of Griffithsin, and (d) purifying the crude extract
by reverse-phase HPLC to obtain Griffithsin (see, also,
Example 1). More specifically, the method involves the use
of ethanol to remove high molecular weight biopolymers from
the extract and the use of an anti-HIV bioassay to guide
fractionation of the extract.
Griffithsin (a polypeptide of exactly SEQ ID NO: 3), which
was isolated and purified using the aforementioned method,
was subjected to conventional procedures typically used to
determine the amino acid sequence of a given pure protein.
Thus, the Griffithsin was initially sequenced by N-terminal
Edman degradation of intact protein and numerous overlapping
peptide fragments generated by endoproteinase digestion.
Amino acid analysis was in agreement with the deduced
sequence. ESI mass spectrometry of reduced, HPLC-purified
Griffithsin showed a molecular ion consistent with the
calculated value. These studies indicated that Griffithsin
from Griffithsia was comprised of a unique sequence of 121
amino acids having little or no significant homology or
identity to previously described proteins or transcription
products of known nucleotide sequences. No more than eight
contiguous amino acids from Griffithsin were found in any
amino acid sequences from known proteins, nor were there any
known proteins from any source having significant sequence
identity with Griffithsin. Given the chemically deduced
amino acid sequence of Griffithsin, a corresponding
recombinant Griffithsin (r-Griffithsin) was created and used
to establish definitively that the deduced amino acid
sequence was, indeed, active against virus, such as HIV and
influenza.
Accordingly, the invention provides isolated and purified
nucleic acid molecules and synthetic nucleic acid molecules,
which comprise a coding sequence for a Griffithsin, such as
an isolated and purified nucleic acid molecule comprising a
sequence of SEQ ID NO: 1, an isolated and purified nucleic
acid molecule encoding an amino acid sequence of SEQ ID NO:
2, an isolated and purified nucleic acid sequence encoding
an amino acid sequence SEQ ID NO: 3, an isolated and
purified nucleic acid molecule comprising a sequence of SEQ
ID NO: 4, an isolated and purified nucleic acid sequence
encoding an amino acid sequence of SEQ ID NO: 5, and a
nucleic acid molecule that is substantially homologous or
substantially identical to any one of the aforementioned
nucleic acid molecules. By "substantially homologous" is
meant sufficient homology to render the polypeptide or
derivative thereof anti-viral, with anti-viral activity
characteristic of an anti-viral protein isolated from
Griffithsia. At least about 50% homology or identity (e.g.,
at least about 60%, at least about 65%, or at least about
70% homology or identity), preferably at least about 75%
homology or identity (e.g., at least about 80% or at least
about 85% homology or identity), and most preferably at
least about 90% homology or identity (e.g., at least about
95% homology or identity) should exist.
The inventive nucleic acid molecule preferably comprises a
nucleic acid sequence encoding at least eight (preferably at
least 10, more preferably at least 20, and most preferably
at least 30) contiguous amino acids of the amino acid
sequence of SEQ ID NO: 3 or SEQ ID NO: 2. The inventive
nucleic acid molecule also comprises a nucleic acid sequence
encoding a polypeptide comprising the amino acid sequence of
a native Griffithsin, in which 1-20, preferably 1-10, more
preferably 1, 2, 3, 4, or 5, and most preferably 1 or 2,
amino acids have been removed from one or both ends,
preferably from only one end, e.g., removed from the
amino-terminal end, of the native Griffithsin.
Alternatively, the nucleic acid molecule can comprise a
nucleic acid sequence encoding a polypeptide comprising the
amino acid sequence of a natural Griffithsin (see SEQ ID NO:
3), in which 1-20, preferably 1-10, more preferably 1, 2, 3,
4, or 5, and most preferably 1 or 2, amino acids have been
added to one or both ends, preferably from only one end,
e.g., the amino-terminal end, of the native Griffithsin.
Preferably, the isolated and purified nucleic acid molecule
encodes a polypeptide comprising at least eight contiguous
amino acids of SEQ ID NO: 3, which desirably have anti-viral
activity. If the at least eight contiguous amino acids
comprise amino acids 1-121 of SEQ ID NO: 3, desirably amino
acids 46, 60, 71, and/or 104 have been rendered
glycosylation resistant, while maintaining antiviral
activity of the polypeptide. Deletions and substitutions of
SEQ ID NO: 2 or SEQ ID NO: 3 are within the skill in the
art.
Given the present disclosure, it will be apparent to one
skilled in the art that a partial Griffithsin gene sequence
will likely suffice to code for a fully functional, i.e.,
anti-viral, such as anti-influenza or anti-HIV, Griffithsin.
A minimum essential DNA coding sequence(s) for a functional
Griffithsin can readily be determined by one skilled in the
art, for example, by synthesis and evaluation of
sub-sequences comprising the native Griffithsin, and by
site-directed mutagenesis studies of the Griffithsin DNA
coding sequence.
Using an appropriate DNA coding sequence, a recombinant
Griffithsin can be made by genetic engineering techniques
(for general background see, e.g., Nicholl, in An
Introduction to Genetic Engineering, Cambridge University
Press: Cambridge (1994), pp. 1-5 & 127-130; Steinberg et
al., in Recombinant DNA Technology Concepts and Biomedical
Applications, Prentice Hall: Englewood Cliffs, N.J. (1993),
pp. 81-124 & 150-162; Sofer in Introduction to Genetic
Engineering, Butterworth-Heinemann, Stoneham, Mass. (1991),
pp. 1-21 & 103-126; Old et al., in Principles of Gene
Manipulation, Blackwell Scientific Publishers: London
(1992), pp. 1-13 & 108-221; and Emtage, in Delivery
Systems for Peptide Drugs, Davis et al., eds., Plenum Press:
New York (1986), pp. 23-33). For example, a Griffithsia gene
or cDNA encoding a Griffithsin can be identified and
subcloned. The gene or cDNA then can be incorporated into an
appropriate expression vector and delivered into an
appropriate polypeptide-synthesizing organism (e.g., E.
coli, S. cerevisiae, P. pastoris, or other bacterial, yeast,
insect, plant or mammalian cells), where the gene, under the
control of an endogenous or exogenous promoter, can be
appropriately transcribed and translated. Alternatively, the
expression vector can be administered to a plant or animal,
for example, for large-scale production (see, e.g., Fischer
et al., Transgenic Res., 9(4-5): 279-299 (2000); Fischer et
al., J. Biol. Regul. Homeost. Agents, 14: 83-92 (2000);
deWilde et al., Plant Molec. Biol., 43: 347-359 (2000);
Houdebine, Transgenic Research, 9: 305-320 (2000); Brink et
al., Theriogenology, 53: 139-148 (2000); Pollock et al., J.
Immunol. Methods, 231: 147-157 (1999); Conrad et al., Plant
Molec. Biol., 38: 101-109 (1998); Staub et al., Nature
Biotech., 18: 333-338 (2000); McCormick et al., PNAS USA,
96: 703-708 (1999); Zeitlin et al., Nature Biotech., 16:
1361-1364 (1998); Tacker et al., Microbes and Infection, 1:
777-783 (1999); Tacket et al., Nature Med., 4(5): 607-609
(1998); and Methods in Biotechnology, Recombinant Proteins
from Plants, Production and Isolation of Clinically Useful
Compounds, Cunningham and Porter, eds., Humana Press:
Totowa, N.J. (1998)). Such expression vectors (including,
but not limited to, phage, cosmid, viral, and plasmid
vectors) are known to those skilled in the art, as are
reagents and techniques appropriate for gene transfer (e.g.,
transfection, electroporation, transduction,
micro-injection, transformation, etc.). If a Griffithsin is
to be recombinantly produced in isolated eukaryotic cells or
in a eukaryotic organism, such as a plant (see above
references and also Methods in Biotechnology, Recombinant
Proteins from Plants, Production and Isolation of Clinically
Useful Compounds, Cunningham and Porter, eds., Humana Press:
Totowa, N.J. (1998)), desirably the N-linked glycosylation
sites at positions 45, 60, 71, and/or 104 is rendered
glycosylation-resistant, such as in accordance with the
methods described herein. Subsequently, the recombinantly
produced polypeptide can be isolated and purified using
standard techniques known in the art (e.g., chromatography,
centrifugation, differential solubility, isoelectric
focusing, etc.), and assayed for anti-viral activity.
Alternatively, a natural Griffithsin can be obtained from
Griffithsia by non-recombinant methods, and sequenced by
conventional techniques. The sequence can then be used to
synthesize the corresponding DNA, which can be subcloned
into an appropriate expression vector and delivered into a
polypeptide-producing cell for en mass recombinant
production of the desired polypeptide.
In this regard, the invention also provides a vector
comprising a DNA sequence, e.g., a Griffithsia gene sequence
for Griffithsin, a cDNA encoding a Griffithsin, or a
synthetic DNA sequence encoding Griffithsin. The vector can
be targeted to a cell-surface receptor if so desired. A
nucleic acid molecule as described above can be cloned into
any suitable vector and can be used to transform or
transfect any suitable host. The selection of vectors and
methods to construct them are commonly known to persons of
ordinary skill in the art and are described in general
technical references (see, in general, "Recombinant DNA Part
D," Methods in Enzymology, Vol. 153, Wu and Grossman, eds.,
Academic Press (1987) and the references cited herein under
"EXAMPLES"). Desirably, the vector comprises regulatory
sequences, such as transcription and translation initiation
and termination codons, which are specific to the type of
host (e.g., bacterium, fungus, plant or animal) into which
the vector is to be introduced, as appropriate and taking
into consideration whether the vector is DNA or RNA.
Preferably, the vector comprises regulatory sequences that
are specific to the genus of the host. Most preferably, the
vector comprises regulatory sequences that are specific to
the species of the host.
Constructs of vectors, which are circular or linear, can be
prepared to contain an entire nucleic acid as described
above or a portion thereof ligated to a replication system
functional in a prokaryotic or eukaryotic host cell.
Replication systems can be derived from ColE1, 2 m[mu]
plasmid, [lambda], SV40, bovine papilloma virus, and the
like.
In addition to the replication system and the inserted
nucleic acid, the construct can include one or more marker
genes, which allow for selection of transformed or
transfected hosts. Marker genes include biocide resistance,
e.g., resistance to antibiotics, heavy metals, etc.,
complementation in an auxotrophic'host to provide
prototrophy, and the like.
One of ordinary skill in the art will appreciate that any of
a number of vectors known in the art are suitable for use in
the invention. Suitable vectors include those designed for
propagation and expansion or for expression or both.
Examples of suitable vectors include, for instance,
plasmids, plasmid-liposome complexes, and viral vectors,
e.g., parvoviral-based vectors (i.e., adeno-associated virus
(AAV)-based vectors), retroviral vectors, herpes simplex
virus (HSV)-based vectors, and adenovirus-based vectors. Any
of these expression constructs can be prepared using
standard recombinant DNA techniques described in, e.g.,
Sambrook et al., Molecular Cloning: A Laboratory Manual,
2edition, Cold Spring Harbor Laboratory Press,
Cold Spring Harbor, N.Y. (1989); Ausubel et al., Current
Protocols in Molecular Biology, Greene Publishing Associates
and John Wiley & Sons, New York, N.Y. (1994); Fischer et
al., Transgenic Res., 9(4-5): 279-299 (2000); Fischer et
al., J. Biol. Regul. Homeost. Agents, 14: 83-92 (2000);
deWilde et al., Plant Molec. Biol., 43: 347-359 (2000);
Houdebine, Transgenic Research, 9: 305-320 (2000); Brink et
al., Theriogenology, 53: 139-148 (2000); Pollock et al., J.
Immunol. Methods, 231: 147-157 (1999); Conrad et al., Plant
Molec. Biol., 38: 101-109 (1998); Staub et al., Nature
Biotech., 18: 333-338 (2000); McCormick et al., PNAS USA,
96: 703-708 (1999); Zeitlin et al., Nature Biotech., 16:
1361-1364 (1998); Tacker et al., Microbes and Infection, 1:
777-783 (1999); and Tacket et al., Nature Med., 4(5):
607-609 (1998). Examples of cloning vectors include the pUC
series, the pBluescript series (Stratagene, LaJolla,
Calif.), the pET series (Novagen, Madison, Wis.), the pGEX
series (Pharmacia Biotech, Uppsala, Sweden), and the pEX
series (Clonetech, Palo Alto, Calif.). Bacteriophage
vectors, such as [lambda]GT10, [lambda]GT11, [lambda]ZapII
(Stratagene), [lambda] EMBL4, and [lambda] NM1149, also can
be used. Examples of plant expression vectors include
pBI101, pBI101.2, pBI101.3, pBI121 and pBIN19 (Clonetech,
Palo Alto, Calif.). Examples of animal expression vectors
include pEUK-C1, pMAM and pMAMneo (Clonetech).
An expression vector can comprise a native or normative
promoter operably linked to an isolated or purified nucleic
acid as described above. The selection of promoters, e.g.,
strong, weak, inducible, tissue-specific and
developmental-specific, is within the skill in the art.
Similarly, the combining of a nucleic acid molecule as
described above with a promoter is also within the skill in
the art.
The DNA, whether isolated and purified or synthetic, or cDNA
encoding a Griffithsin can encode for either the entire
Griffithsin or a portion thereof. Where the DNA or cDNA does
not comprise the entire coding sequence of the native
Griffithsin, the DNA or cDNA can be subcloned as part of a
gene fusion. In a transcriptional gene fusion, the DNA or
cDNA will contain its own control sequence directing
appropriate production of protein (e.g., ribosome binding
site, translation initiation codon, etc.), and the
transcriptional control sequences (e.g., promoter elements
and/or enhancers) will be provided by the vector. In a
translational gene fusion, transcriptional control sequences
as well as at least some of the translational control
sequences (i.e., the translational initiation codon) will be
provided by the vector. In the case of a translational gene
fusion, a chimeric protein will be produced.
Genes also can be constructed for specific fusion proteins
containing a functional Griffithsin component plus a fusion
component conferring additional desired attribute(s) to the
composite protein. For example, a fusion sequence for a
toxin or immunological reagent can be added to facilitate
purification and analysis of the functional protein.
Genes can be specifically constructed to code for fusion
proteins, which contain a Griffithsin coupled to an effector
protein, such as a toxin or immunological reagent, for
specific targeting to a virus or viral-infected cells, e.g.,
HIV and/or HIV-infected cells or influenza and/or
influenza-infected cells. In these instances, the
Griffithsin moiety serves not only as a neutralizing agent
but also as a targeting agent to direct the effector
activities of these molecules selectively against a given
virus, such as HIV or influenza. Thus, for example, a
therapeutic agent can be obtained by combining the
HIV-targeting function or influenza-targeting function of a
functional Griffithsin with a toxin aimed at neutralizing
infectious virus and/or by destroying cells producing
infectious virus, such as HIV or influenza. Similarly, a
therapeutic agent can be obtained, which combines the
viral-targeting function of a Griffithsin with the
multivalency and effector functions of various
immunoglobulin subclasses. Example 6 further illustrates the
viral-targeting, specifically gp120-targeting, properties of
a Griffithsin.
Similar rationales underlie extensive developmental
therapeutic efforts exploiting the HIV gp120-targeting
properties of sCD4. For example, sCD4-toxin conjugates have
been prepared in which sCD4 is coupled to a Pseudomonas
exotoxin component (Chaudhary et al., in The Human
Retrovirus, Gallo et al., eds., Academic Press: San Diego,
Calif. (1991), pp. 379-387; and Chaudhary et al., Nature,
335: 369-372 (1988)), or to a diphtheria toxin component
(Aullo et al., EMBO J., 11: 575-583 (1992)) or to a ricin
A-chain component (Till et al., Science, 242: 1166-1167
(1988)). Likewise, sCD4-immunoglobulin conjugates have been
prepared in attempts to decrease the rate of in vivo
clearance of functional sCD4 activity, to enhance placental
transfer, and to effect a targeted recruitment of
immunological mechanisms of pathogen elimination, such as
phagocytic engulfment and killing by antibody-dependent
cell-mediated cytotoxicity, to kill and/or remove
HIV-infected cells and virus (Capon et al., Nature, 337:
525-531 (1989); Traunecker et al., Nature, 339: 68-70
(1989); and Langner et al. (1993), supra). While such
CD4-immunoglobulin conjugates (sometimes called
"immunoadhesins") have, indeed, shown advantageous
pharmacokinetic and distributional attributes in vivo, and
anti-HIV effects in vitro, clinical results have been
discouraging (Schooley et al. (1990), supra; Husson et al.
(1992), supra; and Langner et al. (1993), supra). This is
not surprising since clinical isolates of HIV, as opposed to
laboratory strains, are highly resistant to binding and
neutralization by sCD4 (Orloff et al. (1995), supra; and
Moore et al. (1992), supra). The Griffithsin polypeptide
binds to a wide range of sugars present on viral
glycoproteins and, therefore, can inhibit a wide range of
viruses which display those glycoproteins. The
extraordinarily broad targeting properties of a functional
Griffithsin to viruses, e.g., primate retroviruses, in
general, and clinical and laboratory strains, in particular,
can be especially advantageous for combining with toxins,
immunoglobulins and other selected effector proteins.
Viral-targeted conjugates can be prepared either by genetic
engineering techniques (see, for example, Chaudhary et al.
(1988), supra) or by chemical coupling of the targeting
component with an effector component. The most feasible or
appropriate technique to be used to construct a given
Griffithsin conjugate or fusion protein will be selected
based upon consideration of the characteristics of the
particular effector molecule selected for coupling to a
Griffithsin. For example, with a selected non-proteinaceous
effector molecule, chemical coupling, rather than genetic
engineering techniques, may be the only feasible option for
creating the desired Griffithsin conjugate.
Accordingly, the invention also provides nucleic acid
molecules encoding Griffithsin fusion proteins. In
particular, the invention provides a nucleic acid molecule
comprising SEQ ID NO: 4 and substantially homologous
sequences thereof. Also provided is a vector comprising a
nucleic acid sequence encoding a Griffithsin fusion protein
and a method of obtaining a Griffithsin fusion protein by
expression of the vector encoding a Griffithsin fusion
protein in a protein-synthesizing organism as described
above. Accordingly, Griffithsin fusion proteins are also
provided.
In view of the above, the invention further provides an
isolated and purified nucleic acid molecule, which comprises
a Griffithsin coding sequence, such as one of the
aforementioned nucleic acids, namely a nucleic acid molecule
encoding an amino acid sequence of SEQ ID NO: 2 or SEQ ID
NO: 3 or a nucleic acid molecule comprising a sequence of
SEQ ID NO: 1 coupled to a second nucleic acid encoding an
effector protein. The first nucleic acid preferably
comprises a nucleic acid sequence encoding at least eight
contiguous amino acids of the amino acid sequence of SEQ ID
NO: 2 or SEQ ID NO: 3, which encodes a functional
Griffithsin, and the second nucleic acid preferably encodes
an effector protein, such as a toxin or immunological
reagent as described herein.
Accordingly, the invention also further provides an isolated
and purified fusion protein encoded by a nucleic acid
molecule comprising a sequence of SEQ ID NO: 1 or a nucleic
acid molecule encoding an amino acid sequence of SEQ ID NO:
2 or SEQ ID NO: 3, either one of which is coupled to a
second nucleic acid encoding an effector protein.
Preferably, the aforementioned nucleic acid molecules encode
at least eight contiguous amino acids of the amino acid
sequence of SEQ ID NO: 2 or SEQ ID NO: 3, which desirably
have anti-viral activity, coupled to an effector molecule,
such as a toxin or immunological reagent as described above.
Preferably, the effector molecule targets a virus, such as
any one or more of the viruses identified herein, especially
an HIV virus, influenza virus (e.g., an H5N1 virus), Severe
Acute Respiratory Syndrome (SARS) virus, Hepatitis C virus,
or Ebola virus. When HIV or influenza is targeted, the
effector molecule preferably targets glycoprotein gp120 of
HIV or hemaglutinin of influenza. If the at least eight
contiguous amino acids of SEQ ID NO: 3 (or SEQ ID NO: 2)
comprise amino acids 1-121, desirably amino acids 46, 60,
71, and/or 104 have been rendered glycosylation-resistant,
yet maintain antiviral activity by substitution of the
asparagine at those positions with, for example, an alanine
or a glutamine residue.
The coupling can be effected at the DNA level or by chemical
coupling as described above. For example, a
Griffithsin-effector protein conjugate of the invention can
be obtained by (a) selecting a desired effector protein or
peptide; (b) synthesizing a composite DNA coding sequence
comprising a first DNA coding sequence comprising one of the
aforementioned nucleic acid sequences, which codes for a
functional Griffithsin, coupled to a second DNA coding
sequence for an effector protein or peptide, e.g., a toxin
or immunological reagent; (c) expressing said composite DNA
coding sequence in an appropriate protein-synthesizing
organism; and (d) purifying the desired fusion protein to
substantially pure form. Alternatively, a
Griffithsin-effector molecule conjugate of the invention can
be obtained by (a) selecting a desired effector molecule and
a Griffithsin or Griffithsin fusion protein; (b) chemically
coupling the Griffithsin or Griffithsin fusion protein to
the effector molecule; and (c) purifying the desired
Griffithsin-effector molecule conjugate to substantially
pure form.
Conjugates comprising a functional Griffithsin (e.g., an
anti-viral polypeptide comprising at least eight contiguous
amino acids of SEQ ID NO: 3, as previously described,
coupled to an anti-Griffithsin antibody, a virus, a viral
glycoprotein, or at least one effector component, which can
be the same or different, such as a toxin, an immunological
reagent, an antiviral agent, or other functional reagent,
can be designed even more specifically to exploit the unique
viral targeting properties of Griffithsins.
Other functional reagents that can be used as effector
components in the inventive conjugates can include, for
example, polyethylene glycol, dextran, albumin, a solid
support matrix, and the like, whose intended effector
functions may include one or more of the following: to
improve stability of the conjugate; to increase the
half-life of the conjugate; to increase resistance of the
conjugate to proteolysis; to decrease the immunogenicity of
the conjugate; to provide a means to attach or immobilize a
functional Griffithsin onto a solid support matrix (e.g.,
see, for example, Harris, in Poly(Ethylene Glycol)
Chemistry: Biotechnical and Biomedical Applications, Harris,
ed., Plenum Press: New York (1992), pp. 1-14). Conjugates
furthermore can comprise a functional Griffithsin coupled to
more than one effector molecule, each of which, optionally,
can have different effector functions (e.g., such as a toxin
molecule (or an immunological reagent) and a polyethylene
glycol (or dextran or albumin) molecule). Diverse
applications and uses of functional proteins and peptides,
such as in the present instance a functional Griffithsin,
attached to or immobilized on a solid support matrix, are
exemplified more specifically for poly(ethylene glycol)
conjugated proteins or peptides in a review by Holmberg et
al. (In Poly(Ethylene Glycol) Chemistry: Biotechnical and
Biomedical Applications, Harris, ed., Plenum Press: New York
(1992), pp. 303-324). Preferred examples of solid support
matrices include magnetic beads, a flow-through matrix, and
a matrix comprising a contraceptive device, such as a
condom, a diaphragm, a cervical cap, a vaginal ring or a
sponge.
Example 4 reveals novel gp120-directed effects of
Griffithsins. Solid-phase ELISA experiments show that
Griffithsin is capable of global conformational effects on
gp120, as observed as a decrease of immunoreactivity at
multiple, distinct, non-overlapping epitopes.
The range of anti-viral activity of Griffithsin against
diverse CD4<+>-tropic immunodeficiency virus strains
in various target cells is remarkable; virtually all tested
strains of HIV-1, HIV-2 and SIV were similarly sensitive to
Griffithsin; clinical isolates and laboratory strains showed
essentially equivalent sensitivity. Cocultivation of
chronically infected and uninfected CEM-SS cells with
Griffithsin did not inhibit viral replication, but did cause
a concentration-dependent inhibition of cell-to-cell fusion
and virus transmission; similar results from binding and
fusion inhibition assays employing
HeLa-CD4-LTR-[beta]-galactosidase cells were consistent with
Griffithsin inhibition of virus-cell and/or cell-cell
binding.
The anti-viral, e.g., anti-HIV, activity of the Griffithsins
and conjugates thereof of the invention can be further
demonstrated in a series of interrelated in vitro anti-viral
assays (Gulakowski et al., J. Virol. Methods, 33: 87-100
(1991)), which accurately predict for anti-viral activity in
humans. These assays measure the ability of compounds to
prevent the replication of HIV and/or the cytopathic effects
of HIV on human target cells. These measurements directly
correlate with the pathogenesis of HIV-induced disease in
vivo. The results of the analysis of the anti-viral activity
of Griffithsins or conjugates, as set forth in Examples 5-7
and 9, predict accurately the anti-viral activity of these
products in vivo in humans and, therefore, establish the
utility of the invention. Furthermore, since the invention
also provides methods of ex vivo use of Griffithsins and
conjugates, the utility of Griffithsins and conjugates
thereof is even more certain.
The Griffithsins and conjugates thereof of the invention can
be used to inhibit a broad range of viruses (see, e.g.,
Principles of Virology: Molecular Biology, Pathogenesis, and
Control, Flint et al., eds., ASM Press: Washington, D.C.
(2000), particularly Chapter 19). Examples of viruses that
may be treated in accordance with the invention include, but
are not limited to, Type C and Type D retroviruses, HTLV-1,
HTLV-2, HIV, FIV, FLV, MLV, BLV, BIV, equine infectious
virus, anemia virus, avian sarcoma viruses, such as Rous
sarcoma virus (RSV), hepatitis type A, B, C, non-A and non-B
viruses, arboviruses, varicella viruses, human herpes virus
(e.g., HHV-6), measles, mumps, filovirus (e.g., Ebola, such
as Ebola strains Sudan, Zaire, Cote d'Ivoire, and Reston),
SARS virus, and rubella viruses. Griffithsins and conjugates
thereof also can be used to inhibit influenza viral
infection, such as an H5N1 viral infection, i.e., a Bird flu
viral infection, (see, e.g., Fields Virology, third edition,
Fields et al., eds., Lippincott-Raven Publishers:
Philadelphia, Pa. (1996), particularly Chapter 45)
prophylactically and therapeutically in accordance with the
methods set forth herein.
The methods, compounds, and compositions described herein
can be used to inhibit any of the foregoing viruses, as well
as other viruses not specifically listed. However, the
methods, compounds, and compositions described herein are
especially useful for the inhibition of the H5N1 virus, SARS
virus, Hepatitis C virus, and Ebola virus as well as
retroviruses such as HIV.
Thus, the invention further provides a composition
comprising (i) one or more of an above-described purified or
isolated nucleic acid or variant thereof, optionally as part
of an encoded fusion protein, and (ii) a carrier, excipient
or adjuvant. Preferably, (i) is present in an antiviral
effective amount and the composition is pharmaceutically
acceptable. The composition can further comprise at least
one additional active agent, such as an antiviral agent
other than a Griffithsin (or antiviral fragment, fusion
protein or conjugate thereof), in an antiviral effective
amount. Suitable antiviral agents include AZT, ddA, ddI,
ddC, 3TC gancyclovir, fluorinated dideoxynucleosides,
acyclovir, [alpha]-interferon, nonnucleoside analog
compounds, such as nevirapine (Shih et al., PNAS, 88:
9878-9882, (1991)), TIBO derivatives, such as R82913 (White
et al., Antiviral Res., 16: 257-266 (1991)), Ro31-8959,
BI-RJ-70 (Merigan, Am. J. Med., 90 (Suppl. 4A): 8S-17S
(1991)), michellamines (Boyd et al., J. Med. Chem., 37:
1740-1745 (1994)) and calanolides (Kashman et al., J. Med.
Chem., 35: 2735-2743 (1992)), nonoxynol-9, gossypol and
derivatives, gramicidin, Enfurtide (i.e., T20), cyanovirin-N
and functional homologs thereof (Boyd et al. (1997), supra).
Other exemplary antiviral compounds include protease
inhibitors (see R. C. Ogden and C. W. Flexner, eds.,
Protease Inhibitors in AIDS Therapy, Marcel Dekker, NY
(2001)), such as saquinavir (see I. B. Duncan and S.
Redshaw, in R. C. Ogden and C. W. Flexner, supra, pp.
27-48), ritonavir (see D. J. Kempf, in R. C. Ogden and C. W.
Flexner, supra, pp. 49-64), indinavir (see B. D. Dorsey and
J. P. Vacca, in R. C. Ogden and C. W. Flexner, supra, pp.
65-84), nelfinavir (see S. H. Reich, in R. C. Ogden and C.
W. Flexner, supra, pp. 85-100), amprenavir (see R. D. Tung,
in R. C. Ogden and C. W. Flexner, supra, pp. 101-118), and
anti-TAT agents. If the composition is to be used to induce
an immune response, it comprises an immune response-inducing
amount of the inventive agent and can further comprise an
immunoadjuvant, such as polyphosphazene polyelectrolyte.
The pharmaceutical composition can contain other
pharmaceuticals, such as virucides, immunomodulators,
immunostimulants, antibiotics and absorption enhancers.
Exemplary immunomodulators and immunostimulants include
various interleukins, sCD4, cytokines, antibody
preparations, blood transfusions, and cell transfusions.
Exemplary antibiotics include antifungal agents,
antibacterial agents, and anti-Pneumocystitis carnii agents.
Exemplary absorption enhancers include bile salts and other
surfactants, saponins, cyclodextrins, and phospholipids
(Davis (1992), supra).
An isolated cell comprising an above-described purified or
isolated nucleic acid or variant thereof, optionally in the
form of a vector, which is optionally targeted to a
cell-surface receptor, is also provided. Examples of host
cells include, but are not limited to, a human cell, a human
cell line, E. coli, B. subtilis, P. aerugenosa, S.
cerevisiae, and N. crassa. E. coli, in particular E. coli
TB-1, TG-2, DH5[alpha], XL-Blue MRF' (Stratagene), SA2821
and Y1090. Preferably, the cell is a mammalian cell,
bacterium, or yeast. A preferred bacterium is lactobacillus
or other commensal microorganism. The above-described
nucleic acid or variant thereof, optionally in the form of a
vector, can be introduced into a host cell using such
techniques as transfection, electroporation, transduction,
micro-injection, transformation, and the like.
Accordingly, the invention provides a method of inhibiting
prophylactically or therapeutically a viral infection, in
particular an influenza viral infection (e.g., an H5N1 viral
infection), a SARS infection, an Ebola infection, a
Hepatitis C viral infection, or HIV infection, of a host.
The method comprises administering to the host an effective
amount of an anti-viral polypeptide, a variant thereof, an
anti-viral polypeptide conjugate or an anti-viral fusion
protein comprising at least eight contiguous amino acids of
SEQ ID NO: 3, wherein the at least eight contiguous amino
acids are nonglycosylated and have anti-viral activity,
whereupon the viral infection is inhibited. The anti-viral
polypeptide can be derived from a Griffithsin obtained from
Griffithsia or recombinantly produced in accordance with the
methods described above. Nonglycosylated anti-viral
polypeptides can be produced in prokaryotic cells/organisms.
Amino acids 45, 60, 71, and/or 104 in such nonglycosylated
antiviral polypeptides can be deleted or substituted, for
example, with alanine or glutamine. Nonglycosylated
antiviral polypeptides also can be produced in eukaryotic
cells/organisms by expressing a portion of a Griffithsin,
such as that of SEQ ID NO: 3, that does not contain a
glycosylation site or all or a portion of a Griffithsin,
such as that of SEQ ID NO: 3, which contains a glycosylation
site that has been rendered glycosylation-resistant as
described and exemplified herein. When the viral infection
is an influenza viral infection and the anti-viral
polypeptide, variant thereof, anti-viral polypeptide
conjugate, or anti-viral fusion protein is administered
topically to the host, preferably the anti-viral protein or
anti-viral peptide is administered to the respiratory system
of the host, preferably as an inhalant, e.g., an inhalant
comprising an aerosol or microparticulate powder.
The prophylactic and therapeutic treatment of many viral
infections, including influenza virus infections, is
complicated by appearance of virus forms resistant to
currently employed medications, such as neuromimidase
inhibitors. The inventive method is particularly useful in
this context, as the inventive anti-viral polypeptide or
anti-viral polypeptide conjugate binds a wide range of
glycoproteins present on the viral surface. Accordingly, the
inventive anti-viral polypeptide, variant, conjugate, or
fusion protein thereof, can be administered to an animal,
preferably a human, dog, cat, bird, cow, pig, horse, lamb,
mouse, or rat, in combination with other anti-viral agents
to guard against the propagation of anti-viral-resistant
strains of virus. In addition, it is thought that during
adaptive mutation (e.g., resistance to neuraminidase
inhibitors), the level of glycosylation found at the viral
surface increases in some viruses, such as influenza. Thus,
in that the inventive anti-viral agent binds sugars of viral
surface glycoproteins, the inventive method provides a
valuable complimentary therapy to current anti-viral
regimens.
Griffithsins and conjugates thereof collectively comprise
polypeptides and proteins, and, as such, are particularly
susceptible to hydrolysis of amide bonds (e.g., catalyzed by
peptidases) and disruption of essential disulfide bonds or
formation of inactivating or unwanted disulfide linkages
(Carone et al., J. Lab. Clin. Med., 100:1-14 (1982)). There
are various ways to alter molecular structure, if necessary,
to provide enhanced stability to the Griffithsin, variant,
fusion protein, or conjugate thereof (Wunsch, Biopolymers,
22: 493-505 (1983); and Samanen, in Polymeric Materials in
Medication, Gebelein et al., eds., Plenum Press: New York
(1985) pp. 227-242), which may be essential for preparation
and use of pharmaceutical compositions containing
Griffithsins, or variants, fusion proteins, or conjugates
thereof, for therapeutic or prophylactic applications
against viruses, e.g., HIV, influenza, (e.g. H5N1),
Hepatitis C, Ebola, or SARS. Possible options for useful
chemical modifications of a Griffithsin, or variant, fusion
protein or conjugate thereof, include, but are not limited
to, the following (adapted from Samanen, J. M. (1985)
supra): (a) olefin substitution, (b) carbonyl reduction, (c)
D-amino acid substitution, (d) N-methyl substitution, (e)
C-methyl substitution, (f) C-C'-methylene insertion, (g)
dehydro amino acid insertion, (h) retro-inverso
modification, (i) N-terminal to C-terminal cyclization, and
(j) thiomethylene modification. Griffithsins, variants,
fusion proteins, and conjugates thereof also can be modified
by covalent attachment of carbohydrate and polyoxyethylene
derivatives, which are expected to enhance stability and
resistance to proteolysis (Abuchowski et al., in Enzymes as
Drugs, Holcenberg et al., eds., John Wiley: New York (1981),
pp. 367-378).
Other important general considerations for design of
delivery strategy systems and compositions, and for routes
of administration, for protein and peptide drugs, such as
Griffithsins, variants, fusion proteins, and conjugates
thereof (Eppstein, CRC Crit. Rev. Therapeutic Drug Carrier
Systems, 5: 99-139 (1988); Siddiqui et al., CRC Crit. Rev.
Therapeutic Drug Carrier Systems, 3: 195-208 (1987); Banga
et al., Int. J. Pharmaceutics, 48: 15-50 (1988); Sanders,
Eur. I Drug Metab. Pharmacokinetics, 15: 95-102 (1990); and
Verhoef, Eur. J. Drug Metab. Pharmacokinetics, 15: 83-93
(1990)), also apply. The appropriate delivery system for a
given Griffithsin, variant, fusion protein, or conjugate
thereof will depend upon its particular nature, the
particular clinical application, and the site of drug
action. As with any protein or peptide drug, oral delivery
of a Griffithsin, variant, fusion protein, or a conjugate
thereof will likely present special problems, due primarily
to instability in the gastrointestinal tract and poor
absorption and bioavailability of intact, bioactive drug
therefrom. Therefore, especially in the case of oral
delivery, but also possibly in conjunction with other routes
of delivery, it will be necessary to use an
absorption-enhancing agent in combination with a given
Griffithsin, variant, fusion protein, or conjugate thereof.
A wide variety of absorption-enhancing agents have been
investigated and/or applied in combination with protein and
peptide drugs for oral delivery and for delivery by other
routes (Verhoef (1990), supra; van Hoogdalem, Pharmac.
Ther., 44: 407-443 (1989); and Davis, J. Pharm. Pharmacol,
44(Suppl. 1): 186-190 (1992)). Most commonly, typical
enhancers fall into the general categories of (a) chelators,
such as EDTA, salicylates, and N-acyl derivatives of
collagen, (b) surfactants, such as lauryl sulfate and
polyoxyethylene-9-lauryl ether, (c) bile salts, such as
glycolate and taurocholate, and derivatives, such as
taurodihydrofusidate, (d) fatty acids, such as oleic acid
and capric acid, and their derivatives, such as
acylcarnitines, monoglycerides and diglycerides, (e)
non-surfactants, such as unsaturated cyclic ureas, (f)
saponins, (g) cyclodextrins, and (h) phospholipids.
Other approaches to enhancing oral delivery of protein and
peptide drugs, such as the Griffithsins, variants, fusion
proteins, and conjugates thereof, can include aforementioned
chemical modifications to enhance stability to
gastrointestinal enzymes and/or increased lipophilicity.
Alternatively, or in addition, the protein or peptide drug
can be administered in combination with other drugs or
substances, which directly inhibit proteases and/or other
potential sources of enzymatic degradation of proteins and
peptides. Yet another alternative approach to prevent or
delay gastrointestinal absorption of protein or peptide
drugs, such as Griffithsins, variants, fusion proteins, or
conjugates thereof, is to incorporate them into a delivery
system that is designed to protect the protein or peptide
from contact with the proteolytic enzymes in the intestinal
lumen and to release the intact protein or peptide only upon
reaching an area favorable for its absorption. A more
specific example of this strategy is the use of
biodegradable microcapsules or microspheres, both to protect
vulnerable drugs from degradation, as well as to effect a
prolonged release of active drug (Deasy, in
Microencapsulation and Related Processes, Swarbrick, ed.,
Marcell Dekker, Inc.: New York (1984), pp. 1-60, 88-89,
208-211). Microcapsules also can provide a useful way to
effect a prolonged delivery of a protein and peptide drug,
such as a Griffithsin, variant, fusion protein, or conjugate
thereof, after injection (Maulding, J. Controlled Release,
6: 167-176 (1987)).
Given the aforementioned potential complexities of
successful oral delivery of a protein or peptide drug, it is
fortunate that there are numerous other potential routes of
delivery of a protein or peptide drug, such as a
Griffithsin, variant, fusion protein, or conjugate thereof.
These routes include topical, subcutaneous, intravenous,
intraarterial, intrathecal, intracisternal, buccal, rectal,
nasal, pulmonary, transdermal, vaginal, ocular, and the like
(Eppstein (1988), supra; Siddiqui et al. (1987), supra;
Banga et al. (1988), supra; Sanders (1990), supra; Verhoef
(1990), supra; Barry, in Delivery Systems for Peptide Drugs,
Davis et al., eds., Plenum Press: New York (1986), pp.
265-275; and Patton et al., Adv. Drug Delivery Rev, 8:
179-196 (1992)). With any of these routes, or, indeed, with
any other route of administration or application, a protein
or peptide drug, such as a Griffithsin, variant, fusion
protein, or conjugate thereof, may initiate an immunogenic
reaction. In such situations it may be necessary to modify
the molecule in order to mask immunogenic groups. It also
can be possible to protect against undesired immune
responses by judicious choice of method of formulation
and/or administration. For example, site-specific delivery
can be employed, as well as masking of recognition sites
from the immune system by use or attachment of a so-called
tolerogen, such as polyethylene glycol, dextran, albumin,
and the like (Abuchowski et al. (1981), supra; Abuchowski et
al., J. Biol. Chem., 252: 3578-3581 (1977); Lisi et al., J.
Appl. Biochem, 4: 19-33 (1982); and Wileman et al., J.
Pharm. Pharmacol, 38: 264-271 (1986)). Such modifications
also can have advantageous effects on stability and
half-life both in vivo and ex vivo.
Procedures for covalent attachment of molecules, such as
polyethylene glycol, dextran, albumin and the like, to
proteins, such as Griffithsins, variants, fusion proteins,
or conjugates thereof, are well-known to those skilled in
the art, and are extensively documented in the literature
(e.g., see Davis et al., in Peptide and Protein Drug
Delivery, Lee, ed., Marcel Dekker: New York (1991), pp.
831-864).
Other strategies to avoid untoward immune reactions also can
include the induction of tolerance by administration
initially of only low doses. In any event, it will be
apparent from the present disclosure to one skilled in the
art that for any particular desired medical application or
use of a Griffithsin or conjugate thereof, the skilled
artisan can select from any of a wide variety of possible
compositions, routes of administration, or sites of
application, what is advantageous.
Accordingly, the anti-viral Griffithsins, variants, fusion
proteins, and conjugates thereof of the invention can be
formulated into various compositions for use, for example,
either in therapeutic treatment methods for infected
individuals, or in prophylactic methods against viral, e.g.,
HIV and influenza virus, infection of uninfected
individuals.
The invention also provides a composition, such as a
pharmaceutical composition, which comprises an isolated and
purified Griffithsin, a variant thereof or fusion protein
comprising the same, a Griffithsin conjugate, a
matrix-anchored Griffithsin or a matrix-anchored Griffithsin
conjugate, such as an anti-viral effective amount thereof.
The composition can further comprise a carrier, such as a
pharmaceutically acceptable carrier. The composition can
further comprise at least one additional anti-viral compound
other than a Griffithsin, variant, fusion protein, or
conjugate thereof, such as in an anti-viral effective amount
of an anti-viral compound. Suitable anti-viral compounds
include cyanovirin, AZT, ddI, ddC, gancyclovir, fluorinated
dideoxynucleosides, nevirapine, R82913, Ro 31-8959,
BI-RJ-70, acyclovir, [alpha]-interferon, recombinant sCD4,
michellamines, calanolides, nonoxynol-9, gossypol and
derivatives thereof, neuroamidase inhibitors, amantatadine,
rimantadine, enfurtide, and gramicidin. If the composition
is to be used to induce an immune response, it comprises an
immune response-inducing amount of a Griffithsin, variant,
fusion protein, or conjugate thereof and can further
comprise an immunoadjuvant, such as polyphosphazene
polyelectrolyte. The Griffithsin used in the composition,
e.g., pharmaceutical composition, can be isolated and
purified from nature or genetically engineered. Similarly,
the Griffithsin conjugate can be genetically engineered or
chemically coupled.
The inventive compositions can be administered to a host,
such as a human, so as to inhibit viral infection in a
prophylactic or therapeutic method. The compositions of the
invention are particularly useful in inhibiting the growth
or replication of a virus, such as influenza virus (e.g., a
H5N1 virus), a SARS virus, an Ebola virus, a Hepatitis C
virus, or a retrovirus, in particular an influenza virus or
an immunodeficiency virus, such as HIV, specifically HIV-1
and HIV-2, inhibiting infectivity of the virus, inhibiting
the binding of virus to a host cell, and the like. The
compositions are useful in the therapeutic or prophylactic
treatment of hosts, e.g., animals, such as humans, who are
infected with a virus or who are at risk for viral
infection, respectively. The compositions also can be used
to treat objects or materials, such as medical equipment,
supplies, or fluids, including biological fluids, such as
blood, blood products and vaccine formulations, cells,
tissues and organs, to remove or inactivate virus in an
effort to prevent or treat viral infection of a host, e.g.,
animal, such as a human. Such compositions also are useful
to prevent sexual transmission of viral infections, e.g.,
HIV, which is the primary way in which the world's AIDS
cases are contracted (Merson (1993), supra). Adherence of
the inventive anti-viral polypeptide, variant, fusion
protein, or conjugate thereof to a solid support, such as a
filter, can be used in clinics to remove all or part of the
viral content of a biological solution. For example, filters
comprising the inventive anti-viral agents can be used to
treat blood supplies prior to transfusion to reduce the risk
of viral transmission. Such filters would find particular
utility in clinics wherein risk of viral infection is high.
It will be appreciated that total removal of the viral
content of a biological solution is not required to achieve
a beneficial effect. Removal of even a fraction of virus
from a biological solution decreases the risk of infection
of a patient.
Potential virucides used or being considered for use against
sexual transmission of HIV are very limited; present agents
in this category include, for example, nonoxynol-9 (Bird,
AIDS, 5: 791-796 (1991)), gossypol and derivatives (Polsky
et al., Contraception, 39: 579-587 (1989); Lin, Antimicrob.
Agents Chemother, 33: 2149-2151 (1989); and Royer,
Pharmacol. Res, 24: 407-412 (1991)), and gramicidin
(Bourinbair, Life Sci./Pharmacol. Lett, 54: PL5-9 (1994);
and Bourinbair et al., Contraception, 49: 131-137 (1994)).
The method of prevention of sexual transmission of viral
infection, e.g., HIV infection, in accordance with the
invention comprises vaginal, rectal, oral, penile or other
topical treatment with an anti-viral effective amount of a
Griffithsin and/or Griffithsin conjugate, alone or in
combination with another anti-viral compound as described
herein.
In a novel approach to anti-HIV prophylaxis pursued under
auspices of the U.S. National Institute of Allergy and
Infectious Diseases (NIAID) (e.g., as conveyed by Painter,
USA Today, Feb. 13, 1996), vaginal suppository instillation
of live cultures of lactobacilli was being evaluated in a
900-woman study. This study was based especially upon
observations of anti-HIV effects of certain H2O2-producing
lactobacilli in vitro (e.g., see published abstract by
Hilier, from NIAID-sponsored Conference on "Advances in AIDS
Vaccine Development," Bethesda, Md., Feb. 11-15, 1996).
Lactobacilli readily populate the vagina, and indeed are a
predominant bacterial population in most healthy women
(Redondo-Lopez et al., Rev. Infect. Dis., 12: 856-872
(1990); Reid et al., Clin. Microbiol. Rev., 3: 335-344
(1990); Bruce and Reid, Can. J. Microbiol., 34: 339-343
(1988); Reu et al., J. Infect. Dis., 171: 1237-1243 (1995);
Hilier et al., Clin. Infect. Dis., 16(Suppl 4): S273-S281;
and Agnew et al., Sex. Transm. Dis., 22: 269-273 (1995)).
Lactobacilli are also prominent, nonpathogenic inhabitants
of other body cavities such as the mouth, nasopharynx, upper
and lower gastrointestinal tracts, and rectum.
It is well-established that lactobacilli can be readily
transduced using available genetic engineering techniques to
incorporate a desired foreign DNA coding sequence, and that
such lactobacilli can be made to express a corresponding
desired foreign protein (see, e.g., Hols et al., Appl. and
Environ. Microbiol., 60: 1401-1413 (1994)). Therefore,
within the context of the present disclosure, it will be
appreciated by one skilled in the art that viable host cells
containing a DNA sequence or vector of the invention, and
expressing a polypeptide, variant, or fusion protein of the
invention, can be used directly as the delivery vehicle for
a Griffithsin, variant, or fusion protein thereof to the
desired site(s) in vivo. Preferred host cells for such
delivery of Griffithsins, variants, or fusion proteins
thereof directly to desired site(s), such as, for example,
to a selected body cavity, can comprise bacteria or yeast.
More specifically, such host cells can comprise suitably
engineered strain(s) of lactobacilli, enterococci, or other
common bacteria; such as E. coli, normal strains of which
are known to commonly populate body cavities. More
specifically yet, such host cells can comprise one or more
selected nonpathogenic strains of lactobacilli, such as
those described by Andreu et al. ((1995), supra), especially
those having high adherence properties to epithelial cells,
such as, for example, adherence to vaginal epithelial cells,
and suitably transformed using the DNA sequences of the
present invention.
As reviewed by McGroarty (FEMS Immunol. Med. Microbiol., 6:
251-264 (1993)) the "probiotic" or direct therapeutic
application of live bacteria, particularly bacteria that
occur normally in nature, more particularly lactobacilli,
for treatment or prophylaxis against pathogenic bacterial or
yeast infections of the urogenital tract, in particular the
female urogenital tract, is a well-established concept.
Recently, the use of a conventional probiotic strategy, in
particular the use of live lactobacilli, to inhibit sexual
transmission of HIV has been suggested, based specifically
upon the normal, endogenous production of virucidal levels
of H2O2 and/or lactic acid and/or other potentially
virucidal substances by certain normal strains of
lactobacilli (e.g., Hilier (1996), supra). However, the
inventive use of non-mammalian cells, particularly bacteria,
more particularly lactobacilli, specifically engineered with
a foreign gene, more specifically a Griffithsin gene, to
express an anti-viral substance, more specifically a
protein, and even more specifically a Griffithsin, is
heretofore unprecedented as a method of treatment of an
animal, specifically a human, to prevent infection by a
virus, specifically a retrovirus, more specifically HIV-1 or
HIV-2.
Elmer et al. (JAMA, 275: 870-876 (1996)) have recently
speculated that "genetic engineering offers the possibility
of using microbes to deliver specific actions or products to
the colon or other mucosal surfaces . . . other fertile
areas for future study include defining the mechanisms of
action of various biotherapeutic agents with the possibility
of applying genetic engineering to enhance activities."
Elmer et al. ((1996), supra) further point out that the
terms "probiotic" and "biotherapeutic agent" have been used
in the literature to describe microorganisms that have
antagonistic activity toward pathogens in vivo; those
authors more specifically prefer the term "biotherapeutic
agent" to denote "microorganisms having specific therapeutic
properties."
In view of the present disclosure, one skilled in the art
will appreciate that the invention teaches an entirely novel
type of "probiotic" or "biotherapeutic" treatment using
specifically engineered strains of microorganisms provided
herein which do not occur in nature. Nonetheless, available
teachings concerning selection of optimal microbial strains,
in particular bacterial strains, for conventional probiotic
or biotherapeutic applications can be employed in the
context of the invention. For example, selection of optimal
lactobacillus strains for genetic engineering,
transformation, direct expression of Griffithsins or
conjugates thereof, and direct probiotic or biotherapeutic
applications, to treat or prevent viral (e.g., HIV)
infection, can be based upon the same or similar criteria,
such as those described by Elmer et al. ((1996), supra),
typically used to select normal, endogenous or
"nonengineered" bacterial strains for conventional probiotic
or biotherapeutic therapy. Furthermore, the recommendations
and characteristics taught by McGroarty, particularly for
selection of optimal lactobacillus strains for conventional
probiotic use against female urogenital infections, are
pertinent to the present invention: " . . . lactobacilli
chosen for incorporation into probiotic preparations should
be easy and, if possible, inexpensive to cultivate . . .
strains should be stable, retain viability following
freeze-drying and, of course, be non-pathogenic to the host
. . . it is essential that lactobacilli chosen for use in
probiotic preparations should adhere well to the vaginal
epithelium . . . ideally, artificially implanted
lactobacilli should adhere to the vaginal epithelium,
integrate with the indigenous microorganisms present, and
proliferate" (McGroarty (1993), supra). While McGroarty's
teachings specifically address selections of "normal"
lactobacillus strains for probiotic uses against pathogenic
bacterial or yeast infections of the female urogenital
tract, similar considerations will apply to the selection of
optimal bacterial strains for genetic engineering and
"probiotic" or "biotherapeutic" application against viral
infections as particularly encompassed by the present
invention.
Accordingly, the method of the invention for the prevention
of sexual transmission of viral infection, e.g., HIV
infection, comprises vaginal, rectal, oral, penile, or other
topical, insertional, or instillational treatment with an
anti-viral effective amount of a Griffithsin, a Griffithsin
conjugate or fusion protein, a matrix-anchored Griffithsin
or conjugate or fusion protein thereof, and/or viable host
cells transformed to express a Griffithsin or conjugate or
fusion protein thereof, alone or in combination with one or
more other anti-viral compound (e.g., as described above).
However, commensal organisms which produce Griffithsin or a
fragment, homolog, or conjugate thereof can inhibit viruses
other than HIV. For example, commensal microorganisms that
produce the inventive polypeptide can be instilled in
mucosal tissue at the site of influenza contact, such as
nasal or oral mucosa, to inhibit influenza infection of a
host.
Compositions for use in the prophylactic or therapeutic
treatment methods of the invention comprise one or more
Griffithsin(s), variant(s), or conjugate(s) or fusion
protein(s) thereof, either one of which can be
matrix-anchored, and desirably a carrier therefor, such as a
pharmaceutically acceptable carrier. Pharmaceutically
acceptable carriers are well-known to those who are skilled
in the art, as are suitable methods of administration. The
choice of carrier will be determined in part by the
particular Griffithsin or conjugate or fusion protein
thereof, as well as by the particular method used to
administer the composition.
One skilled in the art will appreciate that various routes
of administering a drug are available, and, although more
than one route can be used to administer a particular drug,
a particular route can provide a more immediate and more
effective reaction than another route. For example, the
anti-viral agent of the invention can be inhaled in methods
of prophylactically treating a subject for influenza
infection (e.g., an H5N1 infection). Delivery of the
anti-viral agent to a location of initial viral contact,
such as the nose or mouth, blocks the onset of infection.
The anti-viral agent can be administered via subcutaneous
injection. Alternatively, in acute or critical medical
situations, the anti-viral agent can be administered
intravenously. In many cases of infection, a patient
generates an immune response to a virus. However, the
effects of the viral infection so severely compromise the
health of the patient that an effective immune response is
not reached prior to death. Administration of the anti-viral
agent can prolong the life of the patient until a patient's
natural immune defense clears the virus. Furthermore, one
skilled in the art will appreciate that the particular
pharmaceutical carrier employed will depend, in part, upon
the particular Griffithsin or conjugate or fusion protein
thereof employed, and the chosen route of administration.
Accordingly, there is a wide variety of suitable
formulations of the composition of the invention.
Formulations suitable for oral administration can consist of
liquid solutions, such as an effective amount of the
compound dissolved in diluents, such as water, saline, or
fruit juice; capsules, sachets or tablets, each containing a
predetermined amount of the active ingredient, as solid,
granules or freeze-dried cells; solutions or suspensions in
an aqueous liquid; and oil-in-water emulsions or
water-in-oil emulsions. Tablet forms can include one or more
of lactose, mannitol, corn starch, potato starch,
microcrystalline cellulose, acacia, gelatin, colloidal
silicon dioxide, croscarmellose sodium, talc, magnesium
stearate, stearic acid, and other excipients, colorants,
diluents, buffering agents, moistening agents,
preservatives, flavoring agents, and pharmacologically
compatible carriers. Suitable formulations for oral delivery
can also be incorporated into synthetic and natural
polymeric microspheres, or other means to protect the agents
of the present invention from degradation within the
gastrointestinal tract (see, for example, Wallace et al.,
Science, 260: 912-915 (1993)).
The anti-viral agent of the invention (e.g., Griffithsin,
variants, fusion proteins, or conjugates thereof), alone or
in combination with other anti-viral compounds, can be made
into aerosol formulations or microparticulate powder
formulations to be administered via inhalation. These
aerosol formulations can be placed into pressurized
acceptable propellants, such as dichlorodifluoromethane,
propane, nitrogen, and the like.
The anti-viral agent of the invention (e.g., Griffithsin,
variants, fusion proteins, or conjugates thereof), alone or
in combinations with other anti-viral compounds or
absorption modulators, can be made into suitable
formulations for transdermal application and absorption,
such as a patch (Wallace et al. (1993), supra). Transdermal
electroporation or iontophoresis also can be used to promote
and/or control the systemic delivery of the compounds and/or
compositions of the present invention through the skin
(e.g., see Theiss et al., Meth. Find. Exp. Clin. Pharmacol.,
13: 353-359 (1991)).
Formulations suitable for topical administration include
lozenges comprising the active ingredient in a flavor,
usually sucrose and acacia or tragacanth; pastilles
comprising the active ingredient in an inert base, such as
gelatin and glycerin, or sucrose and acacia; and mouthwashes
comprising the active ingredient in a suitable liquid
carrier; as well as creams, emulsions, gels and the like
containing, in addition to the active ingredient, such as,
for example, freeze-dried lactobacilli or live lactobacillus
cultures genetically engineered to directly produce a
Griffithsin, variant, conjugate, or fusion protein thereof
of the present invention, such carriers as are known in the
art. Topical administration is preferred for the
prophylactic and therapeutic treatment of influenza viral
infection, e.g., H5N1 infection, such as through the use of
an inhaler, for example.
Formulations for rectal administration can be presented as a
suppository with a suitable base comprising, for example,
cocoa butter or a salicylate. Formulations suitable for
vaginal administration can be presented as pessaries,
tampons, creams, gels, pastes, foams, or spray formulas
containing, in addition to the active ingredient, such as,
for example, freeze-dried lactobacilli or live lactobacillus
cultures genetically engineered to directly produce a
Griffithsin or conjugate or fusion protein thereof of the
present invention, such carriers as are known in the art to
be appropriate. Similarly, the active ingredient can be
combined with a lubricant as a coating on a condom. Indeed,
preferably, the active ingredient is applied to any
contraceptive device, including, but not limited to, a
condom, a diaphragm, a cervical cap, a vaginal ring, and a
sponge.
Formulations suitable for parenteral administration include
aqueous and non-aqueous, isotonic sterile injection
solutions, which can contain anti-oxidants, buffers,
bacteriostats, and solutes that render the formulation
isotonic with the blood of the intended recipient, and
aqueous and non-aqueous sterile suspensions that can include
suspending agents, solubilizers, thickening agents,
stabilizers, and preservatives. The formulations can be
presented in unit-dose or multi-dose sealed containers, such
as ampules and vials, and can be stored in a freeze-dried
(lyophilized) condition requiring only the addition of the
sterile liquid carrier, for example, water, for injections,
immediately prior to use. Extemporaneous injection solutions
and suspensions can be prepared from sterile powders,
granules, and tablets of the kind previously described.
Formulations comprising a Griffithsin, variant, or fusion
protein thereof, or Griffithsin conjugate suitable for
virucidal (e.g., HIV, Hepatitis C, Ebola, SARS and H5N1)
sterilization of inanimate objects, such as medical supplies
or equipment, laboratory equipment and supplies,
instruments, devices, and the like, can, for example, be
selected or adapted as appropriate, by one skilled in the
art, from any of the aforementioned compositions or
formulations. Preferably, the Griffithsin, variant, or
fusion protein thereof, is produced by recombinant DNA
technology. The Griffithsin conjugate can be produced by
recombinant DNA technology or by chemical coupling of a
Griffithsin, variant, or fusion protein thereof, with an
effector molecule as described above. Similarly,
formulations suitable for ex vivo sterilization,
inactivation, or removal of virus, such as infectious virus,
from a sample, such as blood, blood products, sperm, or
other bodily products, such as a fluid, cells, a tissue or
an organ, or any other solution, suspension, emulsion,
vaccine formulation (such as in the removal of infectious
virus), or any other material which can be administered to a
patient in a medical procedure, can be selected or adapted
as appropriate by one skilled in the art, from any of the
aforementioned compositions or formulations. However,
suitable formulations for ex vivo sterilization or
inactivation or removal of virus from a sample or on an
inanimate object are by no means limited to any of the
aforementioned formulations or compositions. For example,
such formulations or compositions can comprise a functional
Griffithsin, such as that which is encoded by SEQ ID NO: 3,
or anti-viral fragment thereof, such as a fragment
comprising at least eight contiguous amino acids of SEQ ID
NO: 3, wherein the at least eight contiguous amino acids
bind to a virus, or a variant, conjugate, or fusion protein
of either of the foregoing, attached to a solid support
matrix, to facilitate contacting or binding infectious virus
in a sample or removing infectious virus from a sample as
described above, e.g., a bodily product such as a fluid,
cells, a tissue or an organ from an organism, in particular
a mammal, such as a human, including, for example, blood, a
component of blood (e.g., plasma, blood cells, and the
like), or sperm. Preferably, the anti-viral polypeptide
comprises SEQ ID NO: 3. Also preferably, the at least eight
contiguous amino acids bind gp120 of HIV, in particular
infectious HIV. As a more specific example, such a
formulation or composition can comprise a functional
Griffithsin, variant, conjugate or fusion protein thereof,
attached to (e.g., coupled to or immobilized on) a solid
support matrix comprising magnetic beads, to facilitate
contacting, binding and removal of infectious virus, and to
enable magnet-assisted removal of the virus from a sample as
described above, e.g., a bodily product such as a fluid,
cells, a tissue or an organ, e.g., blood, a component of
blood, or sperm. Alternatively, and also preferably, the
solid support matrix comprises a contraceptive device, such
as a condom, a diaphragm, a cervical cap, a vaginal ring, or
a sponge. The anti-viral agent also can be encapsulated or
dispersed within a solid matrix, such as a vaginal ring or
sponge. Methods for encapsulating biotherapeutics into, for
example, biocompatible sustained release devices, are known
in the art.
As an even more specific illustration, such a composition
(e.g., for ex vivo) can comprise a functional (e.g.,
gp120-binding, HIV-inactivating) Griffithsin, variant,
conjugate, or fusion protein thereof, attached to a solid
support matrix, such as magnetic beads or a flow-through
matrix, by means of an anti-Griffithsin antibody or at least
one effector component, which can be the same or different,
such as polyethylene glycol, albumin, or dextran. The
conjugate can further comprise at least one effector
component, which can be the same or different, selected from
the group consisting of, for example, an immunological
reagent and a toxin. A flow-through matrix would comprise,
for instance, a configuration similar to an affinity column.
The Griffithsin can be covalently coupled to a solid support
matrix via an anti-Griffithsin antibody, described below.
Methods of attaching an antibody to a solid support matrix
are well-known in the art (see, for example, Harlow and
Lane. Antibodies: A Laboratory Manual, Cold Springs Harbor
Laboratory: Cold Spring Harbor, N.Y. (1988)). Alternatively,
the solid support matrix, such as magnetic beads, can be
coated with streptavidin, in which case the Griffithsin or
fragment thereof (which comprises at least eight contiguous
amino acids of SEQ ID NO: 3 or SEQ ID NO: 2) or variant
thereof, or a conjugate or fusion protein of either one, is
biotinylated. The at least eight contiguous amino acids of
SEQ ID NO: 2 desirably have anti-viral activity and
preferably bind gp120 of HIV, which preferably is
infectious. Preferably, the anti-viral polypeptide comprises
SEQ ID NO: 3 or SEQ ID NO: 2. Such a composition can be
prepared, for example, by biotinylating the Griffithsin,
variant, conjugate, or fusion protein thereof, and then
contacting the biotinylated protein or peptide with a
(commercially available) solid support matrix, such as
magnetic beads, coated with streptavidin. The use of
biotinylation as a means to attach a desired biologically
active protein or peptide to a streptavidin-coated support
matrix, such as magnetic beads, is well-known in the art.
One skilled in the art will appreciate that a suitable or
appropriate formulation can be selected, adapted or
developed based upon the particular application at hand.
For ex vivo uses, such as virucidal treatments of inanimate
objects or materials, blood or blood products, or tissues,
the amount of Griffithsin, variant, conjugate thereof,
fusion protein thereof, or composition of any of the
foregoing, to be employed should be sufficient that any
virus or virus-producing cells present will be rendered
noninfectious or will be destroyed. For example, for HIV,
this would require that the virus and/or the virus-producing
cells be exposed to concentrations of Griffithsin in the
range of 0.1-1000 nM. Similar considerations apply to in
vivo applications. Therefore, the designation of "anti-viral
effective amount" is used generally to describe the amount
of a particular Griffithsin, variant, conjugate, fusion
protein, or composition thereof required for anti-viral
efficacy in any given application.
In view of the above, the invention also provides a method
of inhibiting prophylactically or therapeutically a viral
infection of a host in which an anti-viral effective amount
of an above-described anti-viral polypeptide, variant,
conjugate, or fusion protein is administered to the host.
Upon administration of the anti-viral effective amount of
the anti-viral polypeptide, variant, conjugate, or fusion
protein, the viral infection is inhibited.
The invention additionally provides a method of
prophylactically or therapeutically inhibiting a viral
infection of a host in which an anti-viral effective amount
of a composition comprising an isolated and purified
anti-viral polypeptide, variant, or anti-viral polypeptide
conjugate or fusion protein, either one of which comprises
at least eight contiguous amino acids of SEQ ID NO: 3 having
anti-viral activity, attached to or encapsulated within a
solid support matrix is administered to the host. By
"therapeutically" is meant that the host already has been
infected with the virus. By "prophylactically" is meant that
the host has not yet been infected with the virus but is at
risk of being infected with the virus. Prophylactic
treatment is intended to encompass any degree of inhibition
of viral infection, including, but not limited to, complete
inhibition, as one of ordinary skill in the art will readily
appreciate that any degree in inhibition of viral infection
is advantageous. Preferably, the inventive active agent is
administered before viral infection or immediately upon
determination of viral infection and is continuously
administered until the virus is undetectable. The method
optionally further comprises the prior, simultaneous or
subsequent administration, by the same route or a different
route, of an antiviral agent or another agent that is
efficacious in inhibiting the viral infection. Upon
administration of the anti-viral effective amount of the
composition, the viral infection is inhibited. Preferably,
the solid support matrix is a contraceptive device, such as
a condom, diaphragm, cervical cap, vaginal ring, or sponge.
In an alternative embodiment, a solid support matrix can be
surgically implanted and later removed.
For in vivo uses, the dose of a Griffithsin, variant,
conjugate, fusion protein, or composition thereof,
administered to an animal, particularly a human, in the
context of the invention should be sufficient to effect a
prophylactic or therapeutic response in the individual over
a reasonable time frame. The dose used to achieve a desired
anti-viral concentration in vivo (e.g., 0.1-1000 nM) will be
determined by the potency of the particular Griffithsin,
variant, fusion protein, or conjugate employed, the severity
of the disease state of infected individuals, as well as, in
the case of systemic administration, the body weight and age
of the infected individual. The size of the dose also will
be determined by the existence of any adverse side effects
that may accompany the particular Griffithsin, variant,
fusion protein, or conjugate or composition thereof,
employed. It is always desirable, whenever possible, to keep
adverse side effects to a minimum.
The invention also provides a method of removing virus, such
as infectious virus, from a sample. The method comprises
contacting the sample with a composition comprising an
isolated and purified anti-viral polypeptide, variant,
conjugate, or fusion protein thereof, comprising at least
eight contiguous amino acids of SEQ ID NO: 3 (or SEQ ID NO:
2). The at least eight contiguous amino acids desirably have
anti-viral activity and bind to the virus and the anti-viral
polypeptide (or conjugate or fusion protein of either of the
foregoing) is attached to a solid support matrix, such as a
magnetic bead. "Attached" is used herein to refer to
attachment to (or coupling to) and immobilization in or on a
solid support matrix. While any means of attachment can be
used, preferably, attachment is by covalent bonds. The
method further comprises separating the sample and the
composition by any suitable means, whereupon the virus, such
as infectious virus, is removed from the sample. Preferably,
the anti-viral polypeptide comprises SEQ ID NO: 3 (or SEQ ID
NO: 2). In one embodiment, the anti-viral polypeptide is
conjugated with an anti-Griffithsin antibody or at least one
effector component, which can be the same or different,
selected from polyethylene glycol, dextran and albumin, in
which case the anti-viral polypeptide is desirably attached
to the solid support matrix through at least one effector
component. The anti-viral polypeptide can be further
conjugated with at least one effector component, which can
be the same or different, selected from the group consisting
of an immunological reagent and a toxin. In another
embodiment, the solid support matrix is coated with
streptavidin and the anti-viral polypeptide is biotinylated.
Through biotin, the biotinylated anti-viral polypeptide is
attached to the streptavidin-coated solid support matrix.
Other types of means, as are known in the art, can be used
to attach a functional Griffithsin (i.e., an anti-viral
polypeptide, variant, conjugate, or fusion protein, as
described above) to a solid support matrix, such as a
magnetic bead, in which case contact with a magnet is used
to separate the sample and the composition. Similarly, other
types of solid support matrices can be used, such as a
matrix comprising a porous surface or membrane, over or
through which a sample is flowed or percolated, thereby
selectively entrapping or removing infectious virus from the
sample. The choice of solid support matrix, means of
attachment of the functional Griffithsin to the solid
support matrix, and means of separating the sample and the
matrix-anchored Griffithsin will depend, in part, on the
sample (e.g., fluid vs. tissue) and the virus to be removed.
It is expected that the use of a selected coupling molecule
can confer certain desired properties to a matrix,
comprising a functional Griffithsin coupled therewith, that
may have particularly advantageous properties in a given
situation. Preferably, the sample is blood, a component of
blood, sperm, cells, tissue or an organ. Also, preferably
the sample is a vaccine formulation, in which case the virus
that is removed is infectious, such as HIV, although HIV, in
particular infectious HIV, can be removed from other samples
in accordance with this method. In another embodiment, the
virus that is removed is a SARS, Ebola, Hepatitis C, or H5N1
virus.
For instance, the skilled practitioner might select a
poly(ethylene glycol) molecule for attaching a functional
Griffithsin to a solid support matrix, thereby to provide a
matrix-anchored Griffithsin, wherein the Griffithsin is
attached to the matrix by a longer "tether" than would be
feasible or possible for other attachment methods, such as
biotinylation/streptavidin coupling. A Griffithsin coupled
by a poly(ethylene glycol) "tether" to a solid support
matrix (such as magnetic beads, porous surface or membrane,
and the like) can permit optimal exposure of a binding
surface, epitope, hydrophobic or electrophilic focus, and/or
the like, on a functional Griffithsin in a manner that, in a
given situation and/or for a particular virus, facilitates
the binding and/or inactivation of the virus. A preferred
solid support matrix is a magnetic bead such that separation
of the sample and the composition is effected by a magnet.
In a preferred embodiment of the method, the at least eight
contiguous amino acids bind gp120 of HIV and HIV is removed
from the sample.
Similarly, other types of solid support matrices can be
used, such as a matrix comprising a porous surface or
membrane, over or through which a sample is flowed or
percolated, thereby selectively inhibiting infectious virus
(e.g., HIV or influenza) in the sample. The choice of solid
support matrix, means of attachment of the functional
Griffithsin to the solid support matrix, and means of
separating the sample and the matrix-anchored Griffithsin
will depend, in part, on the sample (e.g., fluid vs. tissue)
and the virus to be inhibited. It is expected that the use
of a selected coupling molecule can confer certain desired
properties to a matrix, comprising a functional Griffithsin
coupled therewith, that may have particularly advantageous
properties in a given situation.
The methods described herein also have utility in real time
ex vivo inhibition of virus or virus infected cells in a
bodily fluid, such as blood, e.g., in the treatment of viral
infection, or in the inhibition of virus in blood or a
component of blood, e.g., for transfusion, in the inhibition
or prevention of viral infection. Such methods also have
potential utility in dialysis, such as kidney dialysis, and
in inhibiting virus in sperm obtained from a donor for in
vitro and in vivo fertilization. The methods also have
applicability in the context of tissue and organ
transplantations.
In summary, a Griffithsin attached to a solid support
matrix, such as a magnetic bead, can be used to remove
virus, in particular infectious virus, including
immunodeficiency virus, such as HIV, e.g., HIV-1 or HIV-2,
SARS, Ebola, H5N1, and Hepatitis C, from a sample, such as a
sample comprising both infectious and noninfectious virus.
The inventive method also can be used to remove viral
glycoprotein presenting cells, e.g., infected cells that
have, for example, gp120 on their surfaces, from a sample.
The invention, therefore, further provides a composition
comprising naturally-occurring, non-infectious virus, such
as a composition produced as described above. The
composition can further comprise a carrier, such as a
biologically or pharmaceutically acceptable carrier, and an
immuno-adjuvant. Preferably, the noninfectious virus is an
influenza (e.g., H5N1), an Ebola virus, a SARS virus, a
Hepatitis C virus, or an immunodeficiency virus, such as
HIV, e.g., HIV-1 or HIV-2. Alternatively, and also
preferably, the noninfectious virus is FIV. A composition
comprising only naturally-occurring, non-infectious virus
has many applications in research and the prophylactic
treatment of a viral infection. In terms of prophylactic
treatment of a viral infection, the skilled artisan will
appreciate the need to eliminate completely all infectious
virus from the composition. If desired, further treatment of
the composition comprising non-infectious particles with
virus-inactivating chemicals, such as imines or psoralens,
and/or pressure or heat inactivation, will further the
non-infectious nature of the composition. For example, an
immune response-inducing amount of the inventive composition
can be administered to an animal at risk for a viral
infection in order to induce an immune response. The skilled
artisan will appreciate that such a composition is a
significant improvement over previously disclosed
compositions in that the virus is non-infectious and
naturally-occurring. Thus, there is no risk of inadvertent
infection, greater doses can be administered in comparison
to compositions comprising infectious viral particles, and
the subsequent immune response will assuredly be directed to
antigens present on naturally-occurring virus. The
composition comprising naturally-occurring, non-infectious
virus can be administered in any manner appropriate to
induce an immune response. Preferably, the virus is
administered, for example, intramuscularly, mucosally,
intravenously, subcutaneously, or topically. Preferably, the
composition comprises naturally-occurring, non-infectious
human immunodeficiency virus comprising gp120.
The composition comprising naturally-occurring,
non-infectious virus can be combined with various carriers,
adjuvants, diluents or other anti-viral therapeutics, if
desired. Appropriate carriers include, for example,
ovalbumin, albumin, globulins, hemocyanins, and the like.
Adjuvants or immuno-adjuvants are incorporated in most cases
to stimulate further the immune system. Any physiologically
appropriate adjuvant can be used. Suitable adjuvants for
inclusion in the inventive composition include, for example,
aluminum hydroxide, beryllium sulfate, silica, kaolin,
carbon, bacterial endotoxin, saponin, and the like.
Thus, the invention also provides a method of inducing an
immune response to a virus in an animal. The method
comprises administering to the animal an immune
response-inducing amount of a composition comprising
naturally-occurring, non-infectious virus as described
above.
The appropriate dose of a composition comprising
naturally-occurring, non-infectious virus required to induce
an immune response to the virus in an animal is dependent on
numerous factors, such as size of the animal and immune
competency. The amount of composition administered should be
sufficient to induce a humoral and/or cellular immune
response. The amount of non-infectious virus in a particular
composition can be determined using routine methods in the
art, such as the Coulter HIV p24 antigen assay (Coulter
Corp., Hialeah, Fla.). Any suitable dose of a composition
comprising non-infectious virus is appropriate so long as an
immune response is induced, desirably without the appearance
of harmful side effects to the host. In this regard,
compositions comprising from about 10<1 >to about
10<5 >particles, preferably from about 10<2 >to
about 10<4 >particles, most preferably about 10<3
>particles, are suitable for inducing an immune response.
One of ordinary skill can determine the effectiveness of the
composition to induce an immune response using routine
methods known in the art. Cell-mediated response can be
determined by employing, for example, a virus
antigen-stimulated T-cell proliferation assay. The presence
of a humoral immune response can be determined, for
instance, with the Enzyme Linked Immunosorbent Assay
(ELISA). The skilled artisan will appreciate that there are
numerous other suitable assays for evaluating induction of
an immune response. To the extent that a dose is inadequate
to induce an appropriate immune response, "booster"
administrations can subsequently be administered in order to
prompt a more effective immune response.
In terms of administration of the inventive anti-viral
agents or conjugates thereof, the dosage can be in unit
dosage form, such as a tablet or capsule. The term "unit
dosage form" as used herein refers to physically discrete
units suitable as unitary dosages for human and animal
subjects, each unit containing a predetermined quantity of a
Griffithsin or conjugate thereof, alone or in combination
with other anti-viral agents, calculated in an amount
sufficient to produce the desired effect in association with
a pharmaceutically acceptable diluent, carrier, or vehicle.
The specifications for the unit dosage forms of the
invention depend on the particular Griffithsin, variant,
fusion protein, conjugate, or composition thereof, employed
and the effect to be achieved, as well as the
pharmacodynamics associated with each Griffithsin, variant,
fusion protein, conjugate, or composition thereof, in the
host. The dose administered should be an "anti-viral
effective amount" or an amount necessary to achieve an
"effective level" in the individual patient.
Since the "effective level" is used as the preferred
endpoint for dosing, the actual dose and schedule can vary,
depending upon interindividual differences in
pharmacokinetics, drug distribution, and metabolism. The
"effective level" can be defined, for example, as the blood
or tissue level (e.g., 0.1-1000 nM) desired in the patient
that corresponds to a concentration of one or more
Griffithsin or conjugate thereof, which inhibits a virus,
such as HIV, in an assay known to predict for clinical
anti-viral activity of chemical compounds and biological
agents. The "effective level" for agents of the invention
also can vary when the Griffithsin, or conjugate or
composition thereof, is used in combination with AZT or
other known anti-viral compounds or combinations thereof.
One skilled in the art can easily determine the appropriate
dose, schedule, and method of administration for the exact
formulation of the composition being used, in order to
achieve the desired "effective concentration" in the
individual patient. One skilled in the art also can readily
determine and use an appropriate indicator of the "effective
concentration" of the compounds of the invention by a direct
(e.g., analytical chemical analysis) or indirect (e.g., with
surrogate indicators such as p24 or RT) analysis of
appropriate patient samples (e.g., blood and/or tissues).
In the treatment of some virally infected individuals, it
can be desirable to utilize a "mega-dosing" regimen, wherein
a large dose of the Griffithsin or conjugate thereof is
administered, time is allowed for the drug to act, and then
a suitable reagent is administered to the individual to
inactivate the drug.
The pharmaceutical composition can contain other
pharmaceuticals, in conjunction with the Griffithsin,
variant, fusion protein, or conjugate thereof, when used to
therapeutically treat a viral infection, such as an
influenza infection or an HIV infection which results in
AIDS. Representative examples of these additional
pharmaceuticals include anti-viral compounds, virucides,
immunomodulators, immuno stimulants, antibiotics and
absorption enhancers. Exemplary anti-viral compounds include
cyanovirin, AZT, ddI, ddC, gancylclovir, fluorinated
dideoxynucleosides, nonnucleoside analog compounds, such as
nevirapine (Shih et al., PNAS, 88: 9878-9882 (1991)), TIBO
derivatives, such as R82913 (White et al., Anti-viral Res.,
16: 257-266 (1991)), BI-RJ-70 (Merigan, Am. J. Med., 90
(Suppl. 4A): 8S-17S (1991)), michellamines (Boyd et al., J.
Med. Chem., 37: 1740-1745 (1994)) and calanolides (Kashman
et al., J. Med. Chem., 35: 2735-2743 (1992)), nonoxynol -9,
gossypol and derivatives, gramicidin (Bourinbair et al.
(1994), supra), neuraminidase inhibitors, amantadine,
enfurtide, and the like. Exemplary immunomodulators and
immunostimulants include various interleukins, sCD4,
cytokines, antibody preparations, blood transfusions, and
cell transfusions. Exemplary antibiotics include antifungal
agents, antibacterial agents, and anti-Pneumocystitis carnii
agents. Exemplary absorption enhancers include bile salts
and other surfactants, saponins, cyclodextrins, and
phospholipids (Davis (1992), supra).
Administration of a Griffithsin, variant, conjugate, or
fusion protein thereof with other anti-retroviral agents and
particularly with known RT inhibitors, such as ddC, AZT,
ddI, ddA, or other inhibitors that act against other viral,
e.g., HIV, proteins, such as anti-TAT agents, is expected to
inhibit most or all replicative stages of the viral life
cycle. The dosages of ddC and AZT used in AIDS or ARC
patients have been published. A virustatic range of ddC is
generally between 0.05 [mu]M to 1.0 [mu]M. A range of about
0.005-0.25 mg/kg body weight is virustatic in most patients.
The preliminary dose ranges for oral administration are
somewhat broader, for example 0.001 to 0.25 mg/kg given in
one or more doses at intervals of 2, 4, 6, 8, 12, etc.
hours. Currently, 0.01 mg/kg body weight ddC given every 8
hrs is preferred. When given in combined therapy, the other
anti-viral compound, for example, can be given at the same
time as the Griffithsin or conjugate thereof or the dosing
can be staggered as desired. The two drugs also can be
combined in a composition. Doses of each can be less when
used in combination than when either is used alone.
It will also be appreciated by one skilled in the art that a
DNA sequence of a Griffithsin, variant, conjugate, or fusion
protein thereof of the invention can be inserted ex vivo
into mammalian cells previously removed from a given animal,
in particular a human, host. Such cells can be employed to
express the corresponding Griffithsin, variant, conjugate or
fusion protein in vivo after reintroduction into the host.
Feasibility of such a therapeutic strategy to deliver a
therapeutic amount of an agent in close proximity to the
desired target cells and pathogens, i.e., virus, more
particularly SARS, Ebola, Hepatitis C, H5N1, and retrovirus,
specifically HIV and its envelope glycoprotein gp120, has
been demonstrated in studies with cells engineered ex vivo
to express sCD4 (Morgan et al. (1994), supra). It is also
possible that, as an alternative to ex vivo insertion of the
DNA sequences of the invention, such sequences can be
inserted into cells directly in vivo, such as by use of an
appropriate viral vector. Such cells transfected in vivo are
expected to produce anti-viral amounts of Griffithsin,
variant, conjugate, or fusion protein thereof directly in
vivo.
Given the present disclosure, it will be additionally
appreciated that a DNA sequence corresponding to a
Griffithsin, variant, fusion protein, or conjugate thereof
can be inserted into suitable nonmammalian host cells, and
that such host cells will express therapeutic or
prophylactic amounts of a Griffithsin, variant, conjugate,
or fusion protein thereof directly in vivo within a desired
body compartment of an animal, in particular a human.
Example 5 illustrates the transformation and expression of
effective virucidal amounts of a Griffithsin in a
non-mammalian cell, more specifically a bacterial cell. In a
preferred embodiment of the invention, a method of
female-controllable prophylaxis against HIV infection
comprises the intravaginal administration and/or
establishment of, in a female human, a persistent
intravaginal population of lactobacilli that have been
transformed with a coding sequence of the invention to
produce, over a prolonged time, effective virucidal levels
of a Griffithsin, variant, fusion protein, or conjugate
thereof, directly on or within the vaginal and/or cervical
and/or uterine mucosa. It is noteworthy that both the World
Health Organization (WHO), as well as the U.S. National
Institute of Allergy and Infectious Diseases, have pointed
to the need for development of female-controlled topical
microbicides, suitable for blocking the transmission of HIV,
as an urgent global priority (Lange et al., Lancet, 341:
1356 (1993); Fauci, NIAID News, Apr. 27, 1995). A
composition comprising the inventive anti-viral agent and a
solid-support matrix is particularly useful in this regard,
particularly when the solid-support matrix is a
contraceptive device, such as a condom, a diaphragm, a
cervical cap, a vaginal ring, or a sponge. In another
embodiment, a colony of commensal organisms transduced with
the nucleic acid of the invention and producing the
inventive anti-viral agent is applied to mucosal tissue
associated with the onset of influenza infection, such as
respiratory or oral mucosal.
The invention also provides antibodies directed to the
polypeptides of the invention. The availability of
antibodies to any given protein is highly advantageous, as
it provides the basis for a wide variety of qualitative and
quantitative analytical methods, separation and purification
methods, and other useful applications directed to the
subject polypeptides. Accordingly, given the present
disclosure and the polypeptides of the invention, it will be
readily apparent to one skilled in the art that antibodies,
in particular antibodies specifically binding to a
polypeptide of the invention, can be prepared using
well-established methodologies (e.g., such as the
methodologies described in detail by Harlow and Lane, in
Antibodies. A Laboratory Manual, Cold Spring Harbor
Laboratory, Cold Spring Harbor (1988), pp. 1-725). Such
antibodies can comprise both polyclonal and monoclonal
antibodies. Furthermore, such antibodies can be obtained and
employed either in solution-phase or coupled to a desired
solid-phase matrix, such as magnetic beads or a flow through
matrix. Having in hand such antibodies as provided by the
invention, one skilled in the art will further appreciate
that such antibodies, in conjunction with well-established
procedures (e.g., such as described by Harlow and Lane
(1988), supra) comprise useful methods for the detection,
quantification, or purification of a Griffithsin, conjugate
thereof, or host cell transformed to produce a Griffithsin
or conjugate or fusion protein thereof. Example 6 further
illustrates an antibody that specifically binds to a
Griffithsin. Accordingly, the invention further provides a
composition comprising an anti-Griffithsin antibody bound to
the anti-viral agent of the invention, preferably an
anti-viral polypeptide comprising at least eight contiguous
amino acids of SEQ ID NO: 3.
Matrix-anchored anti-Griffithsin antibodies also can be used
in a method to remove virus in a sample. Preferably, the
antibody binds to an epitope of an anti-viral polypeptide of
SEQ ID NO: 2 or SEQ ID NO: 3. Preferably, the matrix is a
solid support matrix, such as a magnetic bead or a
flow-through matrix. If the solid support matrix to which
the anti-Griffithsin antibody is attached comprises magnetic
beads, removal of the antibody-Griffithsin-virus complex can
be readily accomplished using a magnet.
In view of the above, the invention provides a method of
removing virus from a sample. The method comprises (a)
contacting the sample with a composition comprising an
isolated and purified anti-viral polypeptide, variant, or
conjugate or fusion protein thereof, wherein (i) the
anti-viral polypeptide comprises at least eight contiguous
amino acids of SEQ ID NO: 3, and (ii) the at least eight
contiguous amino acids bind to the virus, and (b) contacting
the sample with an anti-Griffithsin antibody attached to a
solid support matrix, whereupon the anti-Griffithsin
antibody binds to the anti-viral polypeptide or conjugate or
fusion protein thereof to which is bound the virus, and (c)
separating the solid support matrix from the sample,
whereupon the virus is removed from the sample. Preferably,
the anti-viral polypeptide comprises SEQ ID NO: 3. The virus
that is removed can be any virus, e.g., a Hepatitis C, SARS,
Ebola, and H5N1 virus. Desirably, the virus that is removed
is infectious, such as HIV, Hepatitis C, Ebola, SARS, and
H5N1. The sample can be blood, a component of blood, sperm,
cells, tissue or an organ.
The antibody for use in the aforementioned method is an
antibody that binds to a polypeptide comprising at least
eight contiguous amino acids of SEQ ID NO: 3, and, which
polypeptide can bind to and inactivate a virus. The antibody
can be coupled to the solid support matrix using similar
methods and with similar considerations as described above
for attaching a Griffithsin to a solid support matrix. For
example, coupling methods and molecules employed to attach
an anti-Griffithsin antibody to a solid support matrix, such
as magnetic beads or a flow-through matrix, can employ
biotin/streptavidin coupling or coupling through molecules,
such as polyethylene glycol, albumin or dextran. Also
analogously, it can be shown that, after such coupling, the
matrix-anchored anti-Griffithsin antibody retains its
ability to bind to a polypeptide comprising at least eight
contiguous amino acids of SEQ ID NO: 3, which polypeptide
can bind to and inactivate a virus.
The invention also provides an anti-Griffithsin antibody
that is anti-idiotypic in respect to a viral glycoprotein,
such as gp120, i.e., has an internal image of gp120 of a
primate immunodeficiency virus. Preferably, the antibody can
compete with gp120 of a primate immunodeficiency virus for
binding to a Griffithsin. In this regard, the primary
immunodeficiency virus preferably is HIV-1 or HIV-2 and the
Griffithsin preferably consists essentially of SEQ ID NO: 2
or SEQ ID NO: 3. Anti-idiotypic antibodies can be generated
in accordance with methods known in the art (see, for
example, Benjamin, in Immunology: a short course,
Wiley-Liss, NY (1996), pp. 436-437; Kuby, in Immunology, 3rd
ed., Freeman, N.Y. (1997), pp. 455-456; Greenspan et al.,
FASEB J., 7: 437-443 (1993); and Poskitt, Vaccine, 9:
792-796 (1991)). Such an anti-idiotypic (in respect to
gp120) anti-Griffithsin antibody is useful in a method of
inhibiting infection of an animal with a virus as provided
herein.
In view of the above, a Griffithsin can be administered to
an animal, the animal generates anti-Griffithsin antibodies,
among which are antibodies that have an internal image of a
viral glycoprotein, such as gp120. In accordance with
well-known methods, polyclonal or monoclonal antibodies can
be obtained, isolated, and selected. Selection of an
anti-Griffithsin antibody that has an internal image of
gp120 can be based upon competition between the
anti-Griffithsin antibody and gp120 for binding to a
Griffithsin, or upon the ability of the anti-Griffithsin
antibody to bind to a free Griffithsin as opposed to a
Griffithsin bound to gp120. Such an anti-Griffithsin
antibody can be administered to an animal to inhibit a viral
infection in accordance with methods provided herein.
Although nonhuman anti-idiotypic antibodies, such as an
anti-Griffithsin antibody that has an internal image of
gp120 and, therefore, is anti-idiotypic to gp120, are
proving useful as vaccine antigens in humans, their
favorable properties might, in certain instances, be further
enhanced and/or their adverse properties further diminished,
through "humanization" strategies, such as those recently
reviewed by Vaughan (Nature Biotech., 16: 535-539 (1998)).
Alternatively, a Griffithsin can be directly administered to
an animal to inhibit a viral infection in accordance with
methods provided herein such that the treated animal,
itself, generates an anti-Griffithsin antibody that has an
internal image of gp120. The production of anti-idiotypic
antibodies, such as anti-Griffithsin antibody that has an
internal image of gp120 and, therefore, is anti-idiotypic to
gp120, in an animal to be treated is known as "anti-idiotype
induction therapy," and is described by Madiyalakan et al.
(Hybridoma, 14: 199-203 (1995)), for example.
In view of the above, the invention enables another method
of inhibiting infection of an animal, such as a mammal, in
particular a human, with a virus. The method comprises
administering to the animal an anti-Griffithsin antibody, or
a composition comprising same, in an amount sufficient to
induce in the animal an immune response to the virus,
whereupon the infection of the animal with the virus is
inhibited. Preferably, the anti-Griffithsin antibody has an
internal image of a viral glycoprotein, such as gp120 of an
immunodeficiency virus with which the animal can be
infected, such as a primate immunodeficiency virus.
Preferably, the antibody can compete with, for example,
gp120 of a primate immunodeficiency virus for binding to a
Griffithsin. In this regard, the primate immunodeficiency
virus preferably is HIV-1 or HIV-2 and the Griffithsin
preferably consists essentially of SEQ ID NO: 3 or SEQ ID
NO: 2. The method can further comprise the administration of
an immunostimulant.
Also enabled by the invention is yet another method of
inhibiting infection of an animal, such as a mammal, in
particular a human, with a virus. The method comprises
administering to the animal a Griffithsin, which binds a
viral glycoprotein, such as gp120 of an immunodeficiency
virus with which the animal can be infected, in an amount
sufficient to induce in the animal an anti-Griffithsin
antibody in an amount sufficient to induce an immune
response to a virus sufficient to inhibit infection of the
animal with the virus. Preferably, the anti-Griffithsin
antibody has an internal image of gp120 of an
immunodeficiency virus with which the animal can be
infected, such as a primate immunodeficiency virus.
Preferably, the antibody can compete with gp120 of a primate
immunodeficiency virus for binding to a Griffithsin. In this
regard, the primate immunodeficiency virus preferably is
HIV-1 or HIV-2 and the Griffithsin preferably consists
essentially of SEQ ID NO: 2 or SEQ ID NO: 3.
With respect to the above methods, sufficient amounts can be
determined in accordance with methods known in the art.
Similarly, the sufficiency of an immune response in the
inhibition of a viral infection in an animal also can be
assessed in accordance with methods known in the art.
Either one of the above methods can further comprise
concurrent, pre- or post-treatment with an adjuvant to
enhance the immune response, such as the prior, simultaneous
or subsequent administration, by the same or a different
route, of an antiviral agent or another agent that is
efficacious in inducing an immune response to the virus,
such as an immunostimulant. See, for example, Harlow et al.
(1988), supra.
The inventive Griffithsins, conjugates, host cells,
antibodies, compositions and methods are further described
in the context of the following examples. These examples
serve to illustrate further the present invention and are
not intended to limit the scope of the invention.
EXAMPLES
Example 1
This example illustrates a method of isolating and purifying
Griffithsin from Griffithsia sp. and elucidating the
Griffithsin amino acid sequence.
Anti-HIV bioassay guided fractionation was used to track the
isolation of the Griffithsin polypeptide. In brief, the
cellular mass from Griffithsia sp. was harvested by
filtration, freeze-dried, and extracted first with H2O
followed by (1:1) MeOH-CH2O2. Individual aliquots of the
organic and aqueous extracts were tested for cytoprotective
properties in the NCI primary anti-HIV screen (Weislow et
al. J. Natl. Cancer Inst., 81: 577-586 (1989)). Only the H2O
extract showed anti-HIV activity.
A freeze-dried aqueous extract (10 g) was brought to a
concentration of 50 mg/ml by addition of DDH2O and
maintained on ice. Crystalline ammonium sulfate (Sigma, St.
Louis, Mo.; molecular biology grade) was added to the
solution such that the final concentration of the mixture
was 75% saturation. The mixture was allowed to precipitate
on ice over night, and was then centrifuged at 3000 rpm for
50 min. The resulting pellets were set aside. The
supernatant was brought to 1 M ammonium sulfate followed by
another round of precipitation and centrifugation. The
pellets from the second centrifugation were saved, and the
resulting supernatant was filtered using a 0.22 [mu]m filter
and subjected to hydrophobic interaction chromatography. A
BioCad workstation (Perseptive Biosystems) was used for the
following column chromatographies. The protein solution from
the centrifugation and filtration steps was injected onto a
Poros PE column (10\*100 mm, Perseptive Biosystems)
pre-equilibrated with a starting buffer of 50 mM sodium
phosphate, 1.5 M ammonium sulfate, pH 7.5. The column was
eluted at a flow rate of 15 ml/min over the following
gradient: (1) 7 column volumes (CV, equal to 7.85 ml) of the
starting buffer; (2) 1.5-0 M ammonium sulfate over 2 CV; (3)
0 M ammonium sulfate for 15 CV. The eluate was monitored for
both conductivity and absorbance (280 nm). Ammonium sulfate
was added to the void fraction possessing anti-HIV activity
to bring the final concentration to 75% saturation. The
mixture was allowed to precipitate on ice overnight, and was
then centrifuged at 3000 rpm for 50 min. DDH2O-resuspended
pellets were first concentrated using a 10 kDa molecular
weight limit membrane, dialyzed against 0.02% sodium azide,
and then brought up to a concentration of 25 mM Tris-HCl, pH
8.5. The resulting protein solution was injected onto a
Poros HQ anion exchange column (10\*100 mm, Perseptive
Biosystems) pre-equilibrated with a starting buffer of 25 mM
Tris-HCl, pH 8.5. The column was eluted at a flow rate of 15
ml/min using the following gradient: (1) 5 CV of the
starting buffer; (2) 0-1 M sodium chloride over 20 CV; (3) 1
M sodium chloride for 5 CV. The eluate was monitored for
absorbance (280 nm). Active fractions from the HQ column
were concentrated and desalted using a 10 kDa molecular
weight limit membrane and subjected to a Bio-RP C4 reverse
phase column (4.6\*100 mm, Covance, Princeton, N.J.) and
eluted at a flow rate of 4 ml/min using the following
gradient: (1) 10 CV of the starting buffer of 5%
acetonitrile in H2O; (2) 5-95% acetonitrile in H2O over 2.5
CV; (3) 95% acetonitrile in H2O for 5 CV. The eluate was
monitored for absorbance (280 nm), and the active fraction
was pooled, lyophilized, and resuspended in
phosphate-buffered saline (PBS), pH 7.4. The protein
solution was injected onto a G3000PW gel permeation column
(21.5\*600 mm, TosoHaas, Montgomeryville, Pa.) and eluted
with PBS, pH 7.4, at a flow rate of 5 ml/min.
Molecular mass and purity (>99%) of Griffithsin were
confirmed by Electrospray ionization mass spectrometry
(ESI-MS), and the protein concentrations were determined by
amino acid analysis. Native molecular weight was determined
by calibrating standard proteins (albumin (68 kDa),
cytochrome c (12.5 kDa), and aprotinin (6.5 kDa)) by their
retention time (as measured by absorbance at 280 nm) and
comparing the resulting calibration curve to the retention
time of the active protein. Amino acid analysis was
accomplished using a Beckman Model 6300 Automated Amino Acid
Analyzer according to manufacturer protocols. N-terminal
amino acid sequencing was performed using an Applied
Biosystems Model 4774A Sequencer according to manufacturer
protocols. Matrix-assisted laser desorption ionization-time
of flight mass spectroscopy (MALDI-TOF MS) was performed
using a Kratos Kompact Maldi III instrument (Shimadzu,
Columbia, Md.) operated in a linear mode using sinapinic
acid as a matrix and trypsin as an external standard. ESI-MS
was performed with a JEOL SX102 equipped with an Analytica
electrospray source. The spectrometer was calibrated using a
lysozyme standard (molecular weight=14305.2) prior to each
analysis. Samples were injected into the source in a 1:1
solution of hexafluorosopropanol and 2% acetic acid. The
masses reported were averages calculated from the various
charged states observed.
Griffithsin was subjected to digestion with cyanogen bromide
(CNBr) and a variety of endoproteinases (Lys-C, Arg-C, and
Asp-N) per manufacturer's instructions. The cleaved peptide
products were purified by reversed-phase HPLC using a
gradient of 0.05% aqueous trifluoroacetic acid for 20 min,
then increasing to 60% acetonitrile in 0.05% aqueous
trifluoroacetic acid over 100 min. Amino acid sequences were
determined by sequential Edman degradation using an Applied
Biosystems Model 494 sequencer according to the protocols of
the manufacturer, and the masses of cleaved peptides were
analyzed by MALDI-TOF mass spectrometer. The amino acid
sequence of the native Griffithsin polypeptide is set forth
as SEQ ID NO: 3.
In summary, the preliminary analysis of the crude aqueous
extract of algae Griffithsia sp. in the NCI's primary in
vitro anti-HIV screening assay (Weislow et al., supra)
identified a protein that bound soluble gp120. The process
described herein is illustrated in FIG. 1. Anti-HIV
bioassay-guided fractionation of the aqueous resulted in the
isolation of Griffithsin. The aqueous extract was subjected
to ammonium sulfate precipitation, hydrophobic interaction
chromatography, anion exchange chromatography,
reversed-phase chromatography, and size exclusion
chromatography to produce a homogeneous protein fraction.
SDS-PAGE analysis showed a single protein band with a
relative molecular mass of approximately 13 kDa, named
Griffithsin. Purified Griffithsin exhibited a single band by
immunoblotting with anti-Griffithsin polyclonal antibodies.
The amino acid sequence of the purified Griffithsin was
established by N-terminal Edman degradation of the intact
protein and by N-terminal sequencing of peptide fragments
cleaved by CNBr and a variety of endopeptidases (Lys-C,
Arg-C, and Asp-N) followed by reversed phase purification
and MALDI-TOF mass spectrometric analysis. The entire 121
amino acid sequence was established except for a single
amino acid at position 31, which does not match any of the
common amino acids. Electrospray ionization mass
spectrometric analysis of isolated Griffithsin showed a
molecular ion with m/z 12,770.05, and the calculated value
for the deduced amino acid sequence without amino acid at
position 31 was m/z 12619.00. It was deduced that the
molecular mass of the amino acid at position 31 was 151.05.
The amino acid analysis of Griffithsin also agreed with the
deduced primary sequence. These data fully support the
proposed primary amino acid sequence of Griffithsin. A
search of the BLAST database (Altschul et al., Nucleic Acids
Res, 25(17), 3389-3402 (1997)) for identification of protein
sequence similarities did not reveal any homologies of
greater than eight contiguous amino acids nor >30% total
sequence homology between Griffithsin and any amino acid
sequences of known proteins or transcription products of
known nucleotide sequences, including the anti-HIV proteins
cyanovirin-N and scytovirin.
Example 2
This example demonstrates the synthesis of Griffithsin
genes. The methods described herein are illustrated in FIG.
2.
The chemically deduced amino acid sequence of Griffithsin
was back-translated to elucidate the corresponding DNA
coding sequence. Since amino acid residue 31 of native
Griffithsin did not appear to be one of the twenty common
amino acids, alanine was substituted in this position (SEQ
ID NO: 2). In order to facilitate initial production and
purification of recombinant Griffithsin, a commercial
expression vector pET-26b(+), from Novagen, Inc., Madison,
Wis., for which reagents were available for affinity
purification and detection, was selected. Appropriate
restriction sites for ligation to pET-26b(+), and a stop
codon, were included in the DNA sequence. SEQ ID NO: 1 is an
example of a DNA sequence encoding a synthetic Griffithsin
gene. A flowchart illustrating a method of synthesizing of a
Griffithsin gene is shown in FIG. 2.
A Griffithsin-encoding DNA sequence was synthesized as 13
overlapping, complementary oligonucleotides and assembled to
form the double-stranded coding sequence. Oligonucleotide
elements of the synthetic DNA coding sequence were
synthesized using a nucleic acid synthesizer (model 394,
Applied Biosystems Inc., Foster City, Calif.). The purified
13 oligonucleotides were individually treated with T4
polynucleotide kinase, and 1 nM quantities of each were
pooled and boiled for 10 minutes to ensure denaturation. The
temperature of the mixture was then reduced to 70[deg.] C.
for annealing of the complementary strands for 15 minutes,
and further reduced to 60[deg.] C. for 15 minutes. The
reaction was cooled on ice and T4 DNA ligase (2,000 units)
additional ligase buffer was added to the reaction. Ligation
of the oligonucleotides was performed with T4 DNA ligase
overnight at 16[deg.] C. The resulting DNA was recovered and
purified from the reaction buffer by phenol:chloroform
extraction, ethanol precipitation, and further washing with
ethanol.
The purified, double-stranded synthetic DNA was then used as
a template in a polymerase chain reaction (PCR). One [mu]l
of the DNA solution obtained after purification of the
ligation reaction mixture was used as a template. Thermal
cycling was performed using a Perkin-Elmer instrument. "Pfu"
thermostable DNA polymerase, restriction enzymes, T4 DNA
ligase, and polynucleotide kinase were obtained from
Stratagene, La Jolla, Calif. Pfu polymerase was selected for
this application because of its claimed superiority in
fidelity compared to the usual Taq enzyme. The PCR reaction
product was run on a 2% agarose gel in TAE buffer. The 465
base pair DNA construct was cut from the gel and purified.
The purified DNA, which was digested with Nde I and Xho I
restriction enzymes, was then ligated into the multicloning
site of the pet-26b(+) vector.
E. coli were transfected with the generated
pET-26b(+)-construct, and recombinant clones were identified
by analysis of restriction digests of plasmid DNA. Sequence
analysis of one of these selected clones indicated that
three bases deviated from the intended coding sequence.
These "mutations," which presumably arose during the PCR
amplification of the synthetic template, were corrected by a
site-directed mutagenesis kit from Stratagene, La Jolla,
Calif. The repair was confirmed by DNA sequence analysis.
For preparation of a DNA sequence encoding a Griffithsin
polypeptide tagged with a penta-His peptide at the
C-terminal end of Griffithsin (e.g., SEQ ID NO: 4), the
aforementioned recombinant Griffithsin construct was
subjected to site-directed mutagenesis to eliminate stop
codons located between the Griffithsin coding sequence and
the penta-His peptide coding sequence using a site-directed
mutagenesis kit from Stratagene, La Jolla, Calif. A pair of
mutagenic oligonucleotide primers were synthesized, which
included portions of the codons encoding the Griffithsin
polypeptide and penta-His peptide, but lacked the stop
codons. Annealing of these mutagenic primers with the
template DNA and extension by DNA polymerase resulted in the
generation of a DNA construct encoding a fusion protein
comprising the Griffithsin amino acid sequence linked to a
penta-His peptide tag. DNA sequencing verified the presence
of the intended sequence.
Example 3
This example demonstrates the expression of an N-terminal
His-tagged-Griffithsin gene.
A recombinant Griffithsin protein and a C-terminal,
His-tagged Griffithsin protein encoded by the nucleic acids
of Example 2 did not efficiently translocate to the
periplasmic fraction of E. coli following protein
expression. In addition, the majority of the produced
proteins accumulated in the inclusion bodies of E. coli
without the cleavage of a pelB signal sequence located at
the N-terminus of the Griffithsin protein. Thus, steps were
taken to express Griffithsin in the cytosolic fraction of E.
coli.
The pET-26b(+)-Griffithsin DNA construct was used as a
template PCR using a pair of appropriate primers. The PCR
product was designed to have a "penta-His" peptide and
thrombin recognition site at the N-terminal end of the
Griffithsin polypeptide, providing for production of a
N-terminal, His-tagged-Griffithsin fusion protein. The PCR
reaction product was purified from an agarose gel. The
purified DNA, which was digested with Nco I and Xho I
restriction enzymes, was ligated into the expression vector
pET-28a(+) vector (Novagen, Inc., Madison, Wis.).
E. coli (strain BL21(DE3)) were transfected with the
pET-28a(+) vector containing the nucleic acid coding
sequence for the His-tagged-Griffithsin fusion protein (see
SEQ ID NO: 4). Selected clones were seeded into small-scale
shake flasks containing LB growth medium with 30 [mu]g/ml
kanamycin and expanded by incubation at 37[deg.] C.
Larger-scale Erlenmeyer flasks (0.5-3.0 liters) were then
seeded. The culture was allowed to grow to a density of
0.5-0.7 OD600 units. The expression of the
His-tagged-Griffithsin fusion protein was induced by adding
IPTG to a final concentration of 1 mM and continuing
incubation at 37[deg.] C. for 3-6 hrs. Bacteria were
harvested by centrifugation, and the soluble fraction was
obtained using BugBuster(TM) reagent and Benzonase nuclease
(Novagen, Inc., Madison, Wis.). Crude soluble fractions
showed both anti-HIV activity and presence of a
His-tagged-Griffithsin fusion protein by Western-blotting.
In addition, the His-tagged-Griffithsin protein accumulated
in the inclusion bodies of E. coli. A flowchart illustrating
a method of expressing and purifying recombinant
His-tagged-Griffithsin is shown in FIG. 3.
The purity (~98%) of recombinant His-tagged Griffithsin was
confirmed by SDS-PAGE on 16% Tricine gel stained by
Coomassie Blue staining. The protein showed the expected
molecular mass for Griffithsin (i.e., 14.6 kDa). Protein
concentrations were determined based on extinction
coefficient at 280 nm of the protein. Approximately 1.6 mg
of recombinant His-tagged Griffithsin was purified from 1 L
of E. coli culture. The purified protein demonstrated
120-binding and anti-viral activity equivalent to that of
native Griffithsin.
This example illustrates a method of producing recombinant
Griffithsin, which displays physical and functional
properties similar, if not identical, to that of natural
Griffithsin.
Example 4
This example describes a method of purifying a recombinant
His-tagged-Griffithsin protein.
Using an immobilized metal affinity chromatography set-up
including Ni-NTA agarose (QIAGEN Inc., Valencia, Calif.), a
His-tagged-Griffithsin fusion protein (as described in
Example 3) was purified.
The soluble fraction described in Example 3 was loaded onto
20 ml gravity columns containing affinity matrix. The
columns were washed extensively with washing buffer (50 mM
NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0) to remove
contaminating proteins. Since His-tagged Griffithsin cannot
compete for binding sites on the Ni-NTA resin if the
imidazole concentration is increased to 100-250 mM, the
His-tagged Griffithsin protein was eluted by applying
elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM
imidazole, pH 8.0) through the column. Column fractions and
wash volumes were monitored by Western-blot analysis using
Penta-His(TM) antibody (QIAGEN Inc., Valencia, Calif.) or
anti-Griffithsin antibody. Fractions containing the purified
His-tagged Griffithsin protein were pooled, dialyzed
extensively against distilled water, and lyophilized.
Potent cytoprotective and anti-replicative activities of
both natural and His-tagged recombinant Griffithsin were
observed using the HIV-1RF strain of HIB in CEM-SS cells.
Both the natural and recombinant Griffithsin polypeptides
demonstrated a concentration-dependent inhibition of
virus-induced cell killing. Griffithsin treatment also
resulted in concomitant decreases in supernatant reverse
transcriptase and viral core antigen, p24. Mid-to-high
picomolar concentrations of Griffithsin exhibited comparably
potent activity against all of the representative T-tropic
laboratory strains and primary isolates as well as M-tropic
primary isolates. In the antiviral assays, there was little
or no evidence of direct cytotoxicity of Griffithsin to the
uninfected control cells at the highest tested
concentrations of Griffithsin (78.3 to 783 nM).
Griffithsin-pretreated uninfected CEM-SS cells retained
normal susceptibility to HIV infection after the removal of
Griffithsin. In contrast, infectivity of cell-free virus was
abolished after pretreatment and removal of Griffithsin.
These results indicate that Griffithsin is a virucide.
Cocultivation of uninfected and chronically infected CEM-SS
with Griffithsin resulted in concentration-dependent
inhibition of cell-cell fusion. Additional binding and
fusion inhibition assay using [beta]-gal indicator cells
showed similar results. Griffithsin inhibited fusion of CD4
[beta]-gal cells with HL 2/3 cells and also inhibited
cell-free HIV-1IIIB fusion and infection of [beta]-gal cells
in a concentration-dependent manner.
Example 5
This example illustrates the anti-HIV activity of natural
Griffithsin polypeptide and His-tagged Griffithsin
polypeptide.
Pure proteins were initially evaluated for antiviral
activity using an XTT-tetrazolium anti-HIV assay described
previously (Boyd, in Aids, Etiology, Diagnosis, Treatment
And Prevention (1988), supra; Gustafson et al., J. Med.
Chem., 35: 1978-1986 (1992); Weislow (1989), supra;
Gulakowski (1991), supra). A CEM-SS human lymphocytic target
cell line was used in all assays maintained in RPMI 1650
medium (Gibco, Grand Island, N.Y.), without phenol red,
supplemented with 5% fetal bovine serum, 2 mM L-Glutamine,
and 50 mg/ml Gentamicin (complete medium).
Exponentially growing cells were pelleted and resuspended at
a concentration of 2.0\*10<5 >cells/ml in complete
medium. The Haitian variant of HIV, HTLV-IIIRF (3.54\*10<6
>SFU/ml), was used throughout. Frozen virus stock
solutions were thawed immediately before use and resuspended
in complete medium to yield 1.2\*10<5 >SFU/ml. The
appropriate amounts of the pure proteins for anti-HIV
evaluations were dissolved in H2O-DMSO (3:1), then diluted
in complete medium to the desired initial concentration. All
serial drug dilutions, reagent additions, and plate-to-plate
transfers were carried out with an automated Biomek 1000
Workstation (Beckman Instruments, Palo Alto, Calif.).
FIG. 4 summarizes the observed antiviral activities of
native Griffithsin from Griffithsia sp. (FIG. 4a) and
recombinant His-tagged-fusion Griffithsin (FIG. 4b). Effects
of a range of concentrations of native Griffithsin and
HIS-tagged-Griffithsin upon CEM-SS cells infected with
HIV-1, as determined after days in culture is illustrated in
FIG. 6. Data points represent the percent of the respective
uninfected, nondrug-treated control values. The two
Griffithsin polypeptides demonstrated potent anti-HIV
activity with an EC50 in the low nanomolar range and no
significant evidence of direct cytotoxicity to the host
cells at the highest tested concentrations (up to 1 mM).
Example 6
This example demonstrates that HIV viral envelope gp120 is
the principal target for Griffithsin.
To determine the affinity of Griffithsin for a series of
protein standards, 100 ng each of gp160, gp120, gp41, sCD4,
bovine IgG, [alpha]-acid glycoprotein, and aprotinin were
subjected to ELISA as previously described (Bokesch et al.,
Biochemistry, 42: 2578-2584 (2003)). Briefly, the protein
standards were bound to a 96-well plate, which was rinsed
with PBST (three times) and blocked with BSA. Between each
step of the protocol, the plate was rinsed with PBST (three
times). The protein standards were incubated with
Griffithsin (100 ng/well), followed by incubation with a
1:500 dilution of an anti-Griffithsin rabbit polyclonal
antibody preparation. Griffithsin bound to the protein
standards was detected by adding goat-anti-rabbit antibodies
conjugated to alkaline phosphatase (Roche Molecular
Biochemicals, Indianapolis, Ind.). Upon addition of alkaline
phosphatase substrate buffer, absorbance was measured at 405
nm for each well. Glycosylation-dependent binding of
Griffithsin to gp120 was examined using an ELISA as above,
with glycosylated and nonglycosylated gp120 (HIV-1SF2 gp120)
added to the 96-well plate and incubated with serial
dilutions of Griffithsin.
Griffithsin was tested for its ability to bind HIV envelope
glycoproteins. Evidence for direct interaction of
Griffithsin with gp120, gp160, and to a lesser degree, gp41
was obtained from ELISA experiments (FIG. 5a). There was
little or no detectable interaction between Griffithsin and
cCD4 or other reference proteins, including bovine IgG,
[alpha]-acid glycoprotein, and aprotinin. An additional
ELISA experiment showed that binding of Griffithsin to
sgp120 is both concentration-dependent and
glycosylation-dependent (FIG. 5b).
To undertake preliminary mapping studies to define
Griffithsin-binding site on the gp120, we evaluated the
effect of Griffithsin on the reactivity of soluble CD4
(sCD4), cyanovirin-N, and a panel of monoclonal antibodies
(mAb) with soluble gp120 (sgp120) in an ELISA format assay.
These studies demonstrated that Griffithsin interfered
strongly with recognition of sgp120 by the mAbs 48d and
2G12. Griffithsin moderately interfered with sCD4 and mAb
IgG 1b12 binding to sgp120. Griffithsin had little or no
effect on the recognition of sgp120 by mAbs that recognize
the C1 region (or V3 loop), and the mAb 17b. However,
additional studies demonstrated that pretreatment of sgp120
with sCD4 and the mAbs IgG1b12, 48d, and 2G12 did not block
subsequent binding of Griffithsin to sgp120. Cyanovirin-N
interfered strongly with the recognition of sgp120 by
Griffithsin. On the other hand, Griffithsin pretreatment of
sgp120 did not block subsequent binding of cyanovirin-N to
sgp120.
Since Griffithsin inhibited viral entry, we compared matched
control and Griffithsin-treated sgp120 preparations in a
flow cytometric sgp120/CD4-expressing cell binding assay to
determine whether Griffithsin inhibits viral attachment or
subsequent fusion events. The CEM-SS cell line expresses
CD4, as demonstrated by the binding of target cells with
both anti-Leu3a and anti-OKT4 monoclonal antibodies. After
incubation of CEM-SS cells with sgp120, the cells were
stained by anti-gp120 mAb-FITC. A concomitant decrease in
the availability of the Leu3a epitope (i.e., the
gp120-binding site on target cells) was observed. In other
words, the sgp120 bound to the gp120 binding site on the
target cells. As expected, little change in the staining
specific for the OKT4 epitope (i.e., a non-gp120 binding
site) was observed. These results are consistent with sgp120
binding of CD4 on the target cells. Pretreatment of sgp120
with Griffithsin substantially recovered the availability of
the Leu3a epitope, indicating that Griffithsin completely
blocked CD4-dependent sgp120 binding. However, overall
sgp120 binding showed two peaks in the flow cytometry data
when Griffithsin-treated sgp120 was added to the cells. The
decreased signal suggests inhibition of sgp120 binding to
CD4 by Griffithsin, which was consistent with the recovery
of the availability of the Leu3a epitope. The increased
signal suggests that the Griffithsin/sgp120 complex also
non-specifically bound to target cells.
This example demonstrates that Griffithsin binds to a region
of gp120 that recognizes CD4 on host cells.
Example 7
This example illustrates the broad-range anti-HIV activity
of Griffithsin.
Anti-viral assays used to study the activities of laboratory
strains and primary isolates of virus have been previously
published (Buckheit et al., Antiviral Res., 21: 247-265
(1993)). The low passage HIV-1 pediatric isolate ROJO was
derived as previously described (Buckheit et al., AIDS Res.
Hum. Retroviruses, 10: 1497-1506 (1994)). Peripheral blood
mononuclear cells (PBMC) and macrophages were isolated from
hepatitis and HIV sero-negative donors following
Ficoll-Hypaque centrifugation as described elsewhere
(Gartner and Popovic, Techniques in HIV Research, Aldovini,
A. and Walker, B., eds., Stockton Press, New York (1994) pp.
59-63). Mean EC50 values were determined from
concentration-response curves from eight dilutions of
Griffithsin (triplicate wells/concentration); assays for
HIV-1 RF/CEM-SS employed XTT-tetrazolium; HIV-1 ROJO were
tested in human PBMC cultures by supernatant reverse
transcriptase activity; HIV-1 Ba-L and ADA were tested in
human primary macrophage cultures by p24 ELISA assay.
Standard errors averaged less than 10% of the respective
means. The results of this study are summarized in Table 1
below.
TABLE 1
Virus Target Cell Tropism EC50 (nM)
HIV-1 Laboratory Strain
RF CEM-SS T 0.043
HIV Primary Isolates
ROJO PBMC T 0.63
ADA Macrophage M 0.50
Ba-L Macrophage M 0.098
The results show that Griffithsin is potently active
(sub-nanomolar EC50 values) against a broad range of HIV
isolates including T-tropic viruses (utilizing CCR5 as a
co-receptor) and M-tropic viruses (utilizing CXCR4 as a
co-receptor). This picomolar level of activity is more
potent than that described for most of the current anti-HIV
agents utilized in therapy or in development, including the
entry inhibitors cyanovirin-N and Enfurtide(R). The data
also show that Griffithsin is effective at inhibiting
infection by both laboratory-adapted strains and, more
importantly, primary clinical isolates of HIV (e.g., ROJO,
ADA, and Ba-L). Finally, the results indicate that
Griffithsin is active regardless of the cell type used in
the assay, having potent activity whether the cells were
T-lymphocytes (CEM-SS), PBMCs, or macrophages. Griffithsin
did not show any toxicity against any of the cell lines even
at concentrations 1000-fold higher than the EC50 values.
Example 8
This example describes the production of anti-Griffithsin
polyclonal antibodies. A flow diagram illustrating a method
of producing anti-Griffithsin antibodies is provided in FIG.
6.
A New Zealand white rabbit was immunized with 100 [mu]g of
Griffithsin in Freund's complete adjuvant. Booster
injections of 50 [mu]g of Griffithsin in Freund's incomplete
adjuvant were administered on days 13, 29, 51, 64, 100, and
195. On days 7, 21, 42, 63, 78, and 112, 10 mL of blood was
removed from the rabbit. On day 112 the rabbit was
sacrificed and bled out. The IgG fraction of the immune sera
of the rabbit was isolated by protein-A Sepharose affinity
chromatography (Bio-Rad, Hercules, Calif.) according to the
manufacturer's instructions. Reactivity of the polyclonal
antibodies for Griffithsin was demonstrated by immunoblot
and ELISA studies with 1:500 to 1:3000 dilution of the
rabbit immunoglobulin fractions.
For immunoblotting, samples were transferred to PVDF
membranes following SDS-PAGE according to standard
procedures. The membranes were incubated for 1 hour with
anti-Griffithsin polyclonal antibodies, washed three times
with PBS containing 0.05% Tween 20 (PBST), and then treated
with goat anti-rabbit IgG antibodies conjugated to
horseradish peroxidase (Sigma, St. Louis, Mo.). After three
washes with PBST, bound antibodies were visualized by
incubating membranes in a solution of 0.05%
3,3'-diaminobenzidine and 0.003% H2O2.
The IgG fraction of rabbit polyclonal anti-Griffithsin
antibodies were purified after the final boost and animal
sacrifice by using protein-A Sepharose chromatography on the
57 mL of rabbit serum collected. Following purification, 78
mL of purified anti-Griffithsin IgGs were produced. The
final concentration of protein was 335 micrograms/mL for a
total yield of 27.3 mg of anti-Griffithsin IgG. To analyze
the specificity of the resulting antibody preparation,
Western blot analysis was performed and resulted in the
clear determination of specificity and avidity for
Griffithsin by the purified antibodies. A 1:250 dilution of
the purified antibodies clearly visualized only the
Griffithsin from a mixture of Griffithsin and other
proteins. The response to Griffithsin by the
anti-Griffithsin antibodies was also shown to be
concentration-dependent.
Example 9
This example illustrates the anti-influenza virus activity
of Griffithsin.
All examined influenza viruses were passaged in Madin Darby
canine kidney (MDCK) cells to prepare viral stocks. MDCK
cells (from ATCC, Manassas, Va.) were grown in
antibiotic-free minimum essential medium (MEM) with
non-essential amino acids (Gibco, Long Island, N.Y.)
containing 5% fetal bovine serum (FBS, HyClone Laboratories,
Logan, Utah) and 0.1% NaHCO3. Test medium consisted of MEM
with 0.18% NaHCO3, 10 units/mL trypsin, 1 [mu]g of
ethylenediaminetetraacetate (EDTA) per ml, and 50 [mu]g
gentamicin/mL.
Inhibition of virus-induced cytopathic effect (CPE) as
determined by visual (microscopic) examination of infected
cells and confirmed by increase in neutral red (NR) dye
uptake into infected cells was used as an indicator of
Griffithsin antiviral activity. The CPE inhibition method
was reported previously by Smee et al. (Antiviral Res., 5:
251-259 (2001)). Seven concentrations of Griffithsin were
screened for antiviral activity against each virus in
96-well flat-bottomed microplates of cells. The Griffithsin
protein was added 5-10 minutes prior to addition of virus to
the cells. The concentration of virus correspond to
approximately 50% infection of cells in culture (CCID50) per
well. The virus challenge dose equals a multiplicity of
infection of approximately 0.001 infectious particles per
cell. The reaction proceeded at 37[deg.] C. for 72 hr. To
perform the NR uptake assay for confirmation of antiviral
activity, dye (0.34% concentration in medium) was added to
the plates used to obtain visual scores of CPE. After 2
hours, color intensity of the dye absorbed by and
subsequently eluted from the cells was determined by the
method of Finter et al., J. Gen. Virol., 5, 419-427 (1969)
using a computerized EL-309 microplate autoreader (Bio-Tek
Instruments, Winooski, Vt.). Antiviral activity was
expressed as the 50% effective (virus-inhibitory)
concentration (EC50 value) determined by plotting
Griffithsin concentration versus percent inhibition on
semi-logarithmic graph paper. Cytotoxicity of compounds was
assessed in parallel with the antiviral determinations in
the same microplates, except in the absence of virus. From
these, 50% cytotoxic endpoints (IC50 values) were
determined. The results of this study are summarized in
Table 2.
TABLE 2
Influenza Virus Strain EC50 ([mu]g/ml)
Beijing/262/95 (H1N1) 0.07
Texas/36/91 (H1N1) 0.06
Los Angeles/2/87 (H3N2) 0.037
Panama/2007/99 (H3N2) 0.006
Shandong/09/93 (H3N2) 0.018
Sydney/5/97 (H3N2) 0.016
Washington/05/96 (H3N2) 0.016
Similar to the results with HIV, Griffithsin was found
to be potently active against a wide spectrum of influenza A
viruses. These viruses included both H1N1 strains and H3N2
strains of influenza, which is especially significant in
light of the fact that the highly virulent Fijian strain of
influenza A that afflicted the United States in 2003/2004
was also a H3N2 strain. Griffithsin was not found to be
toxic to the MDCK cell line utilized for these experiments,
even when the cells were exposed to a high dose of
Griffithsin (10 micrograms/mL).
Example 10
This example describes a method of producing recombinant
Griffithsin.
Recombinant expression of His-tagged Griffithsin in E. coli
was optimized using a fermenter in combination with an
auto-induction media. A seed culture was grown in LB media
containing 30 [mu]g/ml kanamycin in a shaker flask at
37[deg.] C. and 150 rpm for 17 hours. In addition, a
fermenter containing an auto-induction media was inoculated
with the seed culture. The ratio of auto-induction media to
seed culture was approximately 50:1. The culture was grown
at 37[deg.] C. for 24 hours. The final culture density was
approximately 8.6 OD600 units. The final culture was
harvested by centrifugation, and the soluble fraction was
obtained as described above.
Crude soluble fractions contained His-tagged-Griffithsin
fusion protein, which was detected by Western-blotting with
anti-Griffithsin polyclonal antibodies. The ratio of
soluble:insoluble protein at approximately 15 kDa was 50:50.
The ratio indicates that more Griffithsin protein was
produced in soluble fraction in this fermentation procedure
compared with protein expression achieved using a shaker
flask procedure. In addition, the fermentation procedure
provided approximately 30-fold higher quantities of
Griffithsin protein than the shaker flask procedure.
Approximately 50 mg of purified recombinant Griffithsin was
isolated from 1 L of the fermentation. The purified protein
existed as a homodimer and demonstrated gp120 binding and
anti-viral activity equivalent to that of native
Griffithsin.
The results of this example confirm a method of producing
recombinant, anti-viral Griffithsin protein.
Example 11
This example demonstrates the anti-Hepatitis C(HCV) activity
of Griffithsin.
The anti-HCV activity of Griffithsin was analyzed as
generally described in Krieger et al., J. Virol. 75:
4614-4624 (2001), but using the Huh7 ET (luc-ubi-neo/ET)
cell line, which contains a new HCV RNA replicon with a
stable luciferase (LUC) reporter. The HCV RNA replicon ET
contains the 5' N-terminal repeat (IRES) of HCV (5') which
drives the production of a firefly LUC, ubiquitin, and
neomycin phosphotransferase (Neo) fusion protein. Ubiquitin
cleavage releases the LUC and Neo genes. The
encephalomycarditis virus (EMCV) IRES element controls the
translation of the HCV structural proteins NS3-NS5. The NS3
protein cleaves the HCV polyprotein to release the mature
NS3, NS4A, NS4B, NS5A, and NS5B proteins that are required
for HCV replication. At the 3' end of the replicon is the
authentic 3' NTR of HCV. The LUC reporter is used as an
indirect measure of HCV replication. The activity of the LUC
reporter is directly proportional to HCV RNA levels and
positive control antiviral compounds behave comparably using
either LUC or RNA endpoints.
The effect of a His-tagged Griffithsin (SEQ ID NO: 5) added
in triplicate at a single high-test concentration of 20
[mu]M on HCV RNA-derived LUC activity and cytotoxicity was
examined. Human IFN[alpha]-2b was included in each run as a
positive control compound. Subconfluent cultures of the Huh7
ET cell line were plated out into 96-well plates that were
dedicated for the analysis of cell numbers (cytotoxicity) or
antiviral activity and, on the next day, Griffithsin or
IFN[alpha]-2b was added to the appropriate wells. Cells were
processed 72 hr later when the cells were still
subconfluent. Compound cytotoxicity was assessed as the
percent viable cells relative to the untreated cell
controls.
As shown in Table 3, the Griffithsin protein demonstrated
60% inhibition of viral replication, and an overall 25%
reduction in cell growth.
TABLE 3
Inhibition of Viral Cytotoxicity Selective
Activity (% reduction Index (SI)
Compound (% reduction Luc) in rRNA)\*
(IC50/EC50)
Griffithsin 60 74.5 >1
IFN-[alpha] 98.6 98.4 >1
\*Reduction as compared to control cell
The HCV RNA replicon confirmatory assay was then used to
examine the effects of Griffithsin at different
concentrations. Specifically, 0, 0.20, 0.63, 6.32, and 20
[mu]g/ml Griffithsin was tested. Human IFN[alpha]-2b was
included in each run as a positive control compound.
Subconfluent cultures of the Huh7 ET cell line were plated
out into 96-well plates that were dedicated for the analysis
of cell numbers (cytotoxicity) or antiviral activity and, on
the next day, each dose of Griffithsin was added to the
appropriate wells. Cells were processed 72 hr later when the
cells were still subconfluent. Compound EC50 and EC90 values
(antiviral activity) were derived from HCV RNA levels
assessed as either HCV RNA replicon-derived LUC activity or
as HCV RNA using TaqMan RT-PCR. Compound IC50 and IC90
values (cytotoxicity) were calculated where applicable using
CytoTox-1 (Promega), a colorimetric assay used as an
indicator of cell numbers and cytotoxicity when the LUC
assay system was employed, while ribosomal (rRNA) levels
determined via TaqMan RT-PCR were used as an indication of
cell numbers in the RNA-based assay. Compound selectivity
indices SI50 and SI90 values also were calculated from the
spreadsheets.
The results are presented in FIG. 7. As shown in FIG. 7,
Griffithsin demonstrated anti-HCV activity in a
dose-dependent manner. The EC50 of Griffithsin was 7.17
[mu]g/ml indicating substantial anti-viral potency.
Griffithsin never reached a toxicity level, even at the
highest test concentration of 10 [mu]g/ml. Therefore, the
IC50 and IC90 could not be determined.
This example demonstrates that Griffithsin can be used to
effectively inhibit HCV.
Example 12
This example demonstrates the anti-SARS activity of
Griffithsin.
The medium from an 18 h monolayer (80-100% confluent) of
Vero76 cells was drained and a His-tagged Griffithsin (SEQ
ID NO: 5) at 0.1, 0.3, 1.0, 3.2, 10.1, 31.8, or 100 [mu]g/ml
was added, followed within 15 min by the SARS virus or virus
diluent. The plate of treated cells was sealed and incubated
for the standard time period required to induce near-maximal
viral CPE. The plate of cells was then stained with neutral
red as described by Smee et al., Antimicrob. Agents
Chemother. 45: 743-748 (2001) and McManus, Appl.
Environment. Microbiol. 31: 35-38 (1976). Cells not damaged
by virus took up a greater amount of dye. The percentage of
neutral red uptake indicating viable cells was read on a
microplate autoreader at dual wavelengths of 405 and 540 nm,
with the difference taken to eliminate background. An
approximated virus-inhibitory concentration at the 50%
endpoint (EC50) and cell-inhibitory concentration at the 50%
endpoint (IC50) was determined, and a general selectivity
index was calculated from these values: SI=(IC50)/(EC50).
The virus inhibitory EC50 and IC50 values and the SI values
from the neutral red assay are provided in Table 4.
The effect on reduction of virus yield was determined by
assaying frozen and thawed eluates from each cup for virus
titer using serial dilution onto monolayers of susceptible
cells. The development of viral cytopathic effect (CPE) in
these cells was an indication of the presence of infectious
virus. The 90% virus-inhibitory effective concentration
(EC90) of Griffithsin, which is the concentration of
Griffithsin at which the virus yield was inhibited by 1 log
10, was determined from these data. The EC90 value from the
virus yield assay is provided in Table 4.
The visual appearance of treated infected cells was compared
to that of treated uninfected cells. Specifically, changes
such as enlargement, granularity, development of ragged
edges, filmy appearance, rounding, and detachment from the
surface of the well were detected by visual observation.
Based on these observations, the cells were given a
designation of T (100% toxic), PVH (partially toxic-very
heavy-80%), PH (partially toxic-heavy-60%), P (partially
toxic-40%), Ps (partially toxic-slight-20%), or 0 (no
toxicity-0%) conforming to the degree of cytotoxicity
visually present. A 50% virus inhibitory concentration
(EC50) and 50% cell inhibitory (cytotoxic) concentration
(IC50) was determined by regression analysis of these data.
This assay was repeated to confirm the results. The EC50 and
IC50 values and corresponding SI values obtained in the
visual assay and the visual confirmation assay are provided
in Table 4.
TABLE 4
EC50 IC50 EC90
Compound Vehicle Assay ([mu]g/ml)
([mu]g/ml) ([mu]g/ml) SI
Griffithsin water Neutral Red 14
>100 7
water Virus Yield
5 >20
water Visual 4 >100
>25
water Visual- 2 >100
>50
Confirmation
As shown in Table 4, the Griffithsin protein inhibited the
SARS virus with an average EC50 of 3 [mu]g/ml as determined
by visual assay, and an EC90 of 5 [mu]g/ml as determined by
virus yield assay indicating substantial antiviral potency.
The EC50 of 14 [mu]g/ml as determined by the neutral red
assay provides a third measure of anti-viral activity for
this protein. The IC50 of Griffithsin in each assay was
greater than 100 [mu]g/ml, indicating good cell viability at
effective concentrations.
This example demonstrates that Griffithsin can be used to
effectively inhibit the SARS virus.
Example 13
This example demonstrates the anti-H5N1 activity of
Griffithsin.
Madin Darby canine kidney (MDCK) cells, obtained from the
American Type Culture Collection (Manassas, Va.), were grown
in antibiotic-free minimum essential medium with
non-essential amino acids (MEM) (Hyclone Labs, Logan, Utah)
containing 5% fetal bovine serum (FBS) and 0.18% NaHCO3. The
test medium was the above MEM without FBS, with added 10
units/ml trypsin (Sigma, St. Louis, Mo.), 1 [mu]g of
ethylenediaminetetraacetate (EDTA) per ml, and 50 [mu]g
gentamicin/ml. MDCK cells were used for the following cell
culture antiviral studies.
An influenza A (H5N1) hybrid virus was kindly provided by
Medimmune, Inc. (Mountain View, Calif.). The virus consisted
of the core (6 genes) of influenza A/Ann Arbor/6/60 with the
H5 and Ni components from A/Vietnam/1203/2004. The virus was
attenuated and was resistant to amantadine.
Two methods were used to assay the antiviral activity of
His-tagged Griffithsin (SEQ ID NO: 5) against the H5N1 virus
in vitro: inhibition of virus-induced cytopathic effect
(CPE) determined by visual (microscopic) examination of the
cells, and increase in neutral red (NR) dye uptake into
cells, as previously described in Smee et al., Antimicrob.
Agents Chemother. 45: 743-748 (2001).
In the CPE inhibition test, eight concentrations of
Griffithsin or Ribavirin (a positive control; ICN
Pharmaceuticals (Costa Mesa, Calif.)) was added to 96-well
flat-bottomed microplates containing a cell monolayer. The
compound was added 5-10 minutes prior to virus, which was
used at a concentration of approximately 50% cell culture
and 50% infectious doses (CCID50) per well. The viral dose
equated to a multiplicity of infection of approximately
0.001 infectious particles per cell. The plate was sealed
and incubated at 37[deg.] C. The CPE values were read
microscopically after 72 h of incubation. Antiviral activity
expressed as the 50% effective (virus-inhibitory)
concentration (EC50) is provided in Table 5.
The NR assay was performed as reported in Smee et al.,
supra. In the NR uptake assay, dye (0011% final
concentration in medium) was added to the same set of plates
used to obtain the visual scores. After 2 hours, color
intensity of the dye absorbed by and subsequently eluted
from the cells was determined spectrophotometrically.
Antiviral activity was determined by plotting compound
concentration against percent inhibition. The results
expressed as the 50% effective (virus-inhibitory)
concentration (EC50) are provided in Table 5.
Cytotoxicity of compounds was assessed in parallel with the
above antiviral determinations using the same microplates in
the absence of virus. After three days, the percent
inhibition of cell proliferation was assessed by visual and
neutral red assays as described above. From this data, 50%
virus inhibitor concentration (EC50) and 50% cytotoxic
endpoints (IC50) values were determined. Using both
antiviral and cytotoxicity data, selectivity index values
(IC50 divided by EC50) could be calculated for each set of
data. These results are provided in Table 5.
TABLE 5
Visual (CPE) Assay Neutral Red Assay
EC50 IC50 EC50
Compound ([mu]g/ml) ([mu]g/ml) SI
([mu]g/ml) IC50 ([mu]g/ml) SI
Griffithsin 0.65 >10 >15
1.2 >10 >8
Ribavirin 1.8 >100 >56
1.8 >100 >56
As shown in Table 5, Griffithsin demonstrated potent
anti-H5N1 activity, exhibiting an EC50 of 0.65 [mu]g/ml, as
determined by the visual assay, and an EC50 of 1.2 [mu]g/ml,
as determined by the neutral red assay. The IC50 values
demonstrate cytotoxic tolerance at effective concentrations.
This example shows that Griffithsin can be used to
effectively inhibit the anti-H5N1 virus.
Example 14
This example demonstrates the anti-ebola virus activity of
Griffithsin.
Griffithsin is tested for anti-ebola virus activity by using
the Zaire ebola virus engineered to express Green
Fluorescence Protein (GFP), which is described in Towner et
al., Virology. 332(1):20-27 (2005).
A His-tagged Griffithsin (SEQ ID NO: 5) or a positive
control is added to a cell monolayer. Thereafter, the cells
are challenged with the engineered Zaire ebola virus.
Inhibition of ebola viral replication, and thus anti-viral
activity, is detected on the basis of expression of GFP
through means such as flow cytometry analysis. Griffithsin
is found to have anti-ebola virus activity in the low
[mu]g/ml range.
The results indicated that Griffithsin can be used to
effectively inhibit the ebola virus.
The following references, to the extent that they provide
exemplary procedural or other details supplementary to those
set forth herein, are specifically incorporated herein by
reference:
Birren et al., Cold Spring Harbor Laboratory Press, Cold
Spring Harbor, N.Y. (1997),
Birren et al., Genome Analysis: A Laboratory Manual Series,
Volume 2, Detecting Genes, Cold Spring Harbor Laboratory
Press, Cold Spring Harbor, N.Y. (1998),
Birren et al., Genome Analysis: A Laboratory Manual Series,
Volume 3, Cloning Systems, Cold Spring Harbor Laboratory
Press, Cold Spring Harbor, N.Y. (1999),
Birren et al., Genome Analysis: A Laboratory Manual Series,
Volume 4, Mapping Genomes, Cold Spring Harbor Laboratory
Press, Cold Spring Harbor, N.Y. (1999),
Harlow et al., Antibodies: A Laboratory Manual, Cold Spring
Harbor Laboratory Press, Cold Spring Harbor, N.Y. (1988),
Harlow et al., Using Antibodies: A Laboratory Manual, Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
(1999), and
Sambrook et al., Molecular Cloning: A Laboratory Manual, 2nd
edition, Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, N.Y. (1989).
All references, including publications, patent applications,
and patents, cited herein are hereby incorporated by
reference to the same extent as if each reference were
individually and specifically indicated to be incorporated
by reference and were set forth in its entirety herein.
The use of the terms "a" and "an" and "the" and similar
referents in the context of describing the invention
(especially in the context of the following claims) are to
be construed to cover both the singular and the plural,
unless otherwise indicated herein or clearly contradicted by
context. The terms "comprising," "having," "including," and
"containing" are to be construed as open-ended terms (i.e.,
meaning "including, but not limited to,") unless otherwise
noted. Recitation of ranges of values herein are merely
intended to serve as a shorthand method of referring
individually to each separate value falling within the
range, unless otherwise indicated herein, and each separate
value is incorporated into the specification as if it were
individually recited herein. All methods described herein
can be performed in any suitable order unless otherwise
indicated herein or otherwise clearly contradicted by
context. The use of any and all examples, or exemplary
language (e.g., "such as") provided herein, is intended
merely to better illuminate the invention and does not pose
a limitation on the scope of the invention unless otherwise
claimed. No language in the specification should be
construed as indicating any non-claimed element as essential
to the practice of the invention.
Preferred embodiments of this invention are described
herein, including the best mode known to the inventors for
carrying out the invention. Variations of those preferred
embodiments may become apparent to those of ordinary skill
in the art upon reading the foregoing description. The
inventors expect skilled artisans to employ such variations
as appropriate, and the inventors intend for the invention
to be practiced otherwise than as specifically described
herein. Accordingly, this invention includes all
modifications and equivalents of the subject matter recited
in the claims appended hereto as permitted by applicable
law. Moreover, any combination of the above-described
elements in all possible variations thereof is encompassed
by the invention unless otherwise indicated herein or
otherwise clearly contradicted by context.
---
**Scytonema varium red algae
Scytovirin**
**SCYTOVIRIN DOMAIN 1 RELATED POLYPEPTIDES****US8481255****ANTIVIRAL ACTIVITY OF THE PROTEIN SCYTOVIRIN AND METHODS
OF USE****US2011183894****Scytovirins and related conjugates, fusion proteins,
nucleic acids, vectors, host cells, compositions, antibodies
and methods of using scytovirins****US7491798**
---
**Nostoc ellipsosporum****Cyanovirin-N**
**Coded sequence CVN (Cyanovirin-N) mutant with high expression
quantity and high activity and application of coded sequence****CN103255151****CYANOVIRIN VARIANT-POLYMER CONJUGATE****JP4903891****A CYANOVIRIN N MUTANT, MODIFIED DERIVATIVE AND USES
THEREOF****WO2011026351****ANTI-H5N1 INFLUENZA ACTIVITY OF THE ANTIVIRAL PROTEIN
CYANOVIRIN****US2010240578****Antiviral drug combination for livestock****CN101612389****Modified Cyanovirin-N Polypeptide****US2009155304****GENOMIC NUCLEIC ACID SEQUENCE FOR CYANOVIRIN-N AND SIGNAL
PEPTIDE THEREOF****WO2007005766****Anti-cyanovirin antibody****US5998587****Cyanovirin conjugates and matrix-anchored cyanovirin and
related compositions and methods of use****US7105169****Obligate domain-swapped dimer of cyanovirin with enhanced
anti-viral activity****US7276227****Cyanovirin conjugates and matrix-anchored cyanovirin and
related compositions and methods of use****US7048935****Anti-cyanovirin antibody with an internal image of gp120,
a method of use thereof, and a method of using a cyanovirin to
induce an immune response to gp120****US6193982****Cyanovirin conjugates, matrix -anchored cyanovirin and
anti-cyanovirin antibody, and related compositions and methods
of use****AU2003252207**
---**